All published articles of this journal are available on ScienceDirect.

Inflammasome can Affect Adult Neurogenesis: A Review Article
Authors Info & Affiliations
Abstract
Adult neurogenesis is the process of producing new neurons in the adult brain and is limited to two major areas: the hippocampal dentate gyrus and the Subventricular Zone (SVZ). Adult neurogenesis is affected by some physiological, pharmacological, and pathological factors. The inflammasome is a major signalling platform that regulates caspase-1 and induces proinflammatory cytokines production such as interleukin-1β (IL1-β) and IL-18.
Inflammasomes may be stimulated through multiple signals, and some of these signaling factors can affect neurogenesis. In the current review, “adult neurogenesis and inflammasome” were searched in PubMed, Scopus, and Google Scholar. Reviewing various research works showed correlations between inflammasome and neurogenesis by different intermediate factors, such as interferons (IFN), interleukins (IL), α-synuclein, microRNAs, and natural compounds. Concerning the significant role of neurogenesis in the health of the nervous system and memory, understanding factors inducing neurogenesis is crucial for identifying new therapeutic aims. Hence in this review, we will discuss the different mechanisms by which inflammasome influences adult neurogenesis.
1. INTRODUCTION AND STATEMENT OF THE PROBLEM
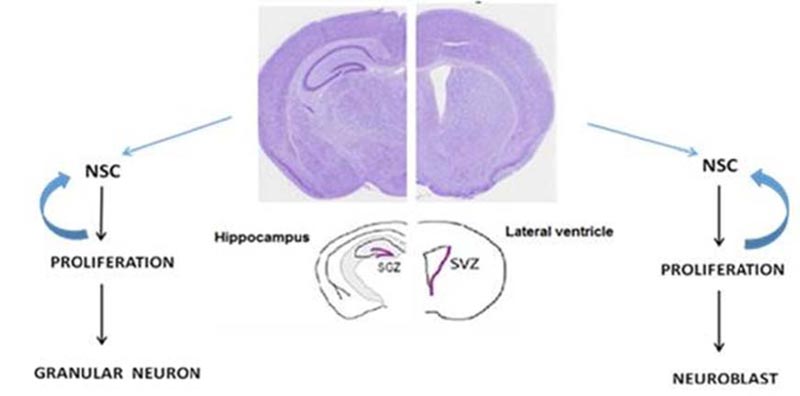
Throughout life, neural stem/progenitor cells [NSPCs] can produce new neurons in specific areas in the mammalian’s brain [1]. Adult neurogenesis contributes to physiological brain activities and is crucial for specific learning and memory processes and a critical factor in adult brain plasticity. Adult neurogenesis is limited to the Subventricular Zone (SVZ) and the hippocampal Dentate Gyrus [2] (Fig. 1).
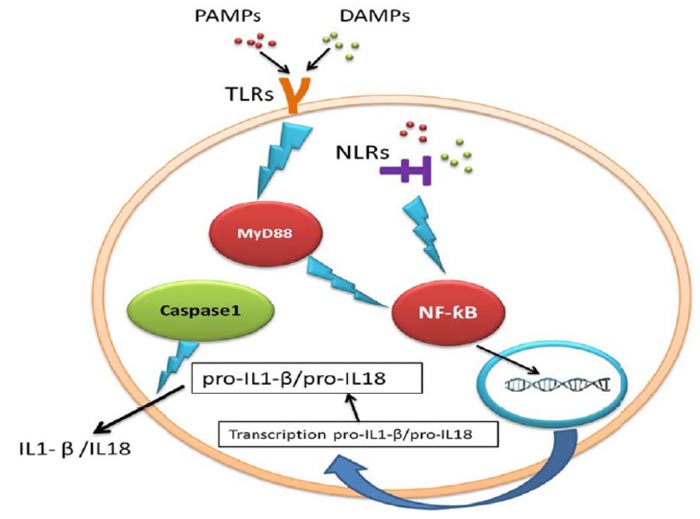
Adult neurogenesis is an active process done with high accuracy and can be affected by pharmacological intervention, different physiological and pathological conditions [3]. Inflammasomes are the main signalling platforms that recognize sterile factors and pathogenic microorganisms [4]. During inflammasome activation, caspase-1, the prominent inflammatory mediator [5], releases interleukin-18 and interleukin-1β (IL-18/ IL1-β), and these products cause pyroptosis and cells death [6, 7]. The innate immune system
cells express Pattern Recognition Receptors (PRRs) to specify two molecular classes: Pathogen-Associated Molecular Patterns (PAMPs) and Damage-Associated Molecular Patterns (DAMPs). NOD-Like Receptors (NLRs) are a subclass of PRRs which are found in the cytosol. There are numerous NLRs proteins that contribute to inflammasome pathways [8]. The oligomerization of NLRs (NLRP1, NLRP3, and NLRC4) can form multi-protein inflammasome complexes [9] (Table 1).
Toll-like Receptors (TLRs) are PRRs on immune cells which detect pathogens and stimulate IL1-β production [10] (Fig. 2). As inflammasome causes pyroptosis, it is possible that inflammasome may reduce neurogenesis. Several studies showed that IL1-β, the product of inflammation and IL-6, and the downstream target of IL1-β, can potentially prevent neurogenesis [2]. IL1-β, a potent pro-inflammatory cytokine, is important for host-defense reactions to injury and infection and is released and produced by many cell types [11]. It was reported that IL1-β had preventive effects on hippocampal cells in the dentate gyrus and NPCs of adult rats [12]. According to the Ryan study, IL1-β reduced neurogenesis and proliferation, while co-treatment with IL1-β receptor antagonist inhibited the adverse impact of IL1-β on the proliferation of cells [13].
| Effect on Neurogenesis | Role in Inflammasome | Factor |
|---|---|---|
| Inhibits neurogenesis | It is produced by activation of the inflammasome | IL1β |
| Inhibits neurogenesis | It is a downstream target of IL1β | IL6 |
| Stimulates neurogenesis | Inhibits IL1β, IL6, and TNF-α | IL4 |
| Stimulates neurogenesis | Inhibits NLRP3 inflammasome | IL10 |
| Inhibits neurogenesis | It is produced by activation of the inflammasome | IL18 |
| Inhibits neurogenesis | Activates inflammasome | INF-α |
| Its role is dose-dependent | In the low dose, it inhibits the activation of NLRP1 inflammasome. Activates inflammasome at high doses |
INF- β |
| Stimulates neurogenesis | Inhibits α-synuclein and inhibits NLRP3 inflammasome | miR-7 |
| Stimulates neurogenesis | Inhibits NLRP3 inflammasome | miR-9 |
| Stimulates neurogenesis | Inhibits NLRP3 inflammasome | miR-223 |
| Stimulates neurogenesis | Inhibits TXNIP that is one activator of NLRP3 inflammasome | Curcumin |
| Stimulates neurogenesis | Inhibits NLRP3 inflammasome | EGCG |
| Stimulates neurogenesis | Inhibits NLRP3 inflammasome | Quercetin |
| Stimulates neurogenesis | Inhibits NLRP3 inflammasome | Boswellia serrate |
IL1-β also decreased proliferation and differentiation of rat neonatal DG NPCs to serotonergic (5-hydroxytryptamine) neurons [14]. Since IL-1 β is the inflammasome's ultimate production, therefore neurogenesis may be enhanced by managing the inflammasome and reducing the IL1-β production.
Interleukin-6 (IL-6) is an IL1-β downstream target, increased continuously in patient’s serum with NLRP3 inflammasome mediated conditions [2]. IL-6 reduced neurogenesis and enhanced apoptosis in rat adult DG NPCs, blocking antibodies to IL-6 induced neurogenesis [15].
The crucial role of IL-18 is highlighted in mediating neurodegeneration and neuroinflammation in the CNS under pathological conditions [16]. IL-18 exhibits direct pro-inflammatory properties by increasing inflammatory factors such as TNF-α, IL1-β, and IL-6 [17]. As previously described, TNF-α, IL1-β, and IL-6 have inhibitory effects on neurogenesis. Also, IL-18 increases amyloid precursor protein [18], Beta-secretase 1 (BACE1), and the N-terminal fragment of presenilin-1 and presenilin enhancer-2 protein levels [19]. It has been reported that in the adult SGZ, presenilin (PS) variant expression is correlated to early-onset familial Alzheimer's [20]. So, IL-18 secreted during inflammasome influences neurogenesis not only by above mentioned inflammatory factors, but also via presenilin expression in adult SGZ.
Another interleukin that, unlike the expressed interleukins, has a protective effect against inflammation is IL-4. It inhibits inflammasome and reduces IL-6, IL1-β, and TNF-α [21]. In many studies, the effect of IL-4 on immunity has been shown. It is also prominent in the normal brain’s functions, involving learning, memory, and neurogenesis [22]. IL-4 increases neuronal and glial differentiation [14]. Changes in microglial status by IL-4 alter their phenotype to enhance neurogenesis [23, 24]. It can be said that IL-4 enhances neurogenesis via inhibiting inflammasome, IL1-β, TNF-α, and IL-6 and therefore increases neurogenesis. IL-10 also inhibits NLRP3 inflammasome and increases neurogenesis through the inhibition of IL1-β production [25]. It elevates proliferation and decreases neuronal and glial differentiation [14]. According to the Gurung et al. study, IL-10 controls NLRP3 inflammasome activation negatively [26]. It inhibits the signaling of nuclear factor kappa B [NF-κB] during inflammasome [27]. It also prevents TNF-α generation of human monocytes [28]. In neurons, this interleukin receptor signaling is correlated with elevated cell survival [29] and the regulation of adult neurogenesis [30].
Another effective factor in the inflammasome pathway is TLR. TLRs, as the main regulators of the innate immune system, are receptors that contributed to the inflammasome. The immune signal is activated through NF-κB to prime the NLRP3 inflammasome [31].
IFN-α and IFN-β are inflammasome products and control inflammasome activation [32].
Interferons α and β, in addition to being key players at the start of the inflammasome, are two of its ultimate products [33]. It has been assumed that IFN-α suppresses NSC proliferation directly and, as a result, reduces new neurons generation [34] and inhibits the proliferation of cells in adult rat’s SGZ [35]. Chronic peripheral IFN-α administration suppresses neurogenesis and induces depressive behavioral phenotypes [34]. The impact of IFN-β on neurogenesis depends on its concentration. Low IFN-β concentrations (1000 U/ml) contribute to the survival and proliferation of human NSC, while greater concentrations (more than 100,000 U/ml) reduce the survival and proliferation of human NSC [36]. Controlling IFN-α and IFN-β from various pathways, like inflammasome, can enhance neurogenesis. There are other factors that cause neurogenesis suppression by activating the inflammation. α-synuclein (α-syn) is a ubiquitous protein particularly observed in high amounts in the brain and is assumed to have the main role in the pathogenesis of Alzheimer’s and Parkinson’s disease and other neurodegenerative disorders [37]. In patients with dementia, levels of hippocampal α-syn have been reported to increase [38]. α-syn motivates IL1-β production in a process that is dependent on the NLRP3 inflammasome in monocytes [39]. A53T mutant α-syn inhibited proliferation and differentiation of adult NSC in SVZ through activation of NLRP3/caspase-1/IL1-β signalling axis. Extracellular α-syn may stimulate pathways of pro-inflammatory TLR4 in astrocytes [40]. TLR4 generates IL1-β by the activation of the inflammasome. IL1-β inhibits neurogenesis. A negative effect of α-syn has been detected on newly generated neurons, especially on their dendrite development and spine improvement [38]. Accumulation of α-syn may be involved in the neurodegenerative process via weakening neurogenesis and the existence of differentiating neural progeny [41]. It can be summarized that α-syn causes an inhibitory effect on neurogenesis via activation of the inflammasome. Studies have shown that some microRNA inhibits TLR3 and NLRP3 by hindering α-synuclein. MicroRNA7, microRNA223, and microRNA9 inhibit the activation of NLRP3 inflammasome in NSCs, resulting in increased neurogenesis [42-45]. miR-7 regulates NLRP3 and TLR4 expression in adult NSCs directly [46]. miR-7 inhibits the activation of the microglial NLRP3 inflammasome, while anti-miR-7 aggravates the activation of inflammasome in vitro [42]. miR-7 simulator’s injection into lateral ventricles prevented the activation of NLRP3 inflammasome and enhanced adult neurogenesis in mouse SVZ [46]. Furthermore, intracerebroventricular injection of miR-7 improved the SVZ neurogenesis impairment and promoted adult SVZ neurogenesis by reducing NLRP3 inflammasome [46].
It was shown that miR-223 reduced the NLRP3 activity [43, 44] and cell-autonomous suppression of miR-223 in the adult mice dentate gyrus NS/PCs caused a potential rise in immature neurons soma size, dendritic tree size, branch number per neuron, and complexity; however, neuronal migration in the dentate gyrus was not affected [45]. miR-9 can also prevent the NLRP3 inflammasome activation [47] and control NS/PC proliferation [45]. Levels of miR-9 expression increase across the transition from neuronal precursors to neurons during the differentiation of embryonic stem cells [48]. miR-7, miR-9, and miR-223 may increase neurogenesis by suppression of inflammasome.
Some natural compounds have stimulant impacts on neurogenesis [49-51]. Curcumin [52], Epigallocatechin-3-gallate (EGCG), Boswellia serrate [53], and quercetin are examples of these natural compounds that enhance neurogenesis. Curcumin can decrease the release of IL1-β from microglia, inhibit microglial activation, decrease stroke injury, activate neurogenesis in the hippocampus, and trigger neuronal protective mechanisms like heat shock protein [HSP] elevation [54]. Studies reported that HSP90 inhibition influences the stability of free NLRP3 and pro-IL1-β proteins [55]. Epigallocatechin-3-gallate [EGCG] is the major antioxidant in green tea. EGCG, by activating SHH and suppressing NLRP3 and NF-κB, inhibits IL1-β, TNF-α, and caspase1 and stimulates neurogenesis [56]. Tozser et al. showed that EGCG promotes hippocampal adult neurogenesis [57]. Treatment of EGCG markedly increased BrdU-labeled cells in hippocampal cultures, neural progenitor cell (NPC), and in the adult mice’s dentate gyrus. Other findings showed that EGCG prompts neurogenesis via reduction of IL1-β and SHH pathway and, SHH ameliorates neurogenesis by the suppression of the inflammation pathway.
Boswellia serrate resin extract, which has 3-O-acetyl-11-keto-b-boswellic acid (AKBA), possesses anti-inflammatory features and has significant therapeutic properties to cure inflammatory illnesses. It decreases IL1-β and TNF-α in a dose-dependent manner [58]. It also controls inflammasome, decreases inflammatory cytokines, and suppresses NF-κB p65, TLR-3, and TLR-4 protein expression [59]. Boswellic acid inhibits TNF-α and IL1-β release in monocytes [60] and inhibits IL-1, IL-6, TNFα, and NF-κB [61]. Hippocampal neurogenesis after administration of Boswellia extracts significantly increased [53]. Boswellia Serrata up-regulates Brain-Derived Neurotrophic Factor [BDNF]. BDNF regulates adult SVZ neurogenesis [61]. The role played by BDNF in controlling SVZ neurogenesis was primarily observed by Goldman et al. in the 1990s. They reported that rat SVZ-derived neuroblasts treated with BDNF in vitro survived for a long time [62].
CONCLUSION
It is concluded that activation of the inflammasome in the brain can inhibit adult neurogenesis, and drugs or compounds that inhibit the inflammasome platform can improve the neurogenesis process in adults by different pathways.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICTS OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

