All published articles of this journal are available on ScienceDirect.

Migraine: An Overview
Abstract
The pathophysiology of migraine is not completely understood and continues to be investigated. The complexity of interactions taking place in the sensory neuronal network with the mediation of all different neurotransmitters involved ives the measure of the extreme difficulty connected with the knowledge of migraine pathogenesis and in particular of its cardinal sign. Neuronal components are relevant in migraine pathophysiology: there could be a generalized interictal abnormal excitability of the cerebral cortex in migraine, possibly favoring the occurrence of spreading depression with consequent activation of the trigeminal system. Many theories have been formulated in these last sixty years about the pathogenesis of migraine and other forms of primary headache, but the problem is still far to be fully clarified. The present review is focused on the description of different theories on the migraine pathogenesis.
INTRODUCTION
Migraine is a neurological syndrome characterized by altered perceptions, headaches, and nausea. Migraine is an invalidating disturbance largely spread (>12%) in the world population, having the form of an autonomous disease, as well secondary or indirect (2-3%), in this last case being only a pathologic symptom of different etiologies. The last headache classification, published in 2004, divides, respectively, in four and ten groups, with relative subtypes, the primary and secondary headaches [1]. The typical migraine headache is unilateral, throbbing, moderate to severe. The pain may be bilateral at the onset or start on one side and become generalized, and usually alternates sides from one attack to the next. The pain peaks and then subsides, and usually lasts between 4 and 72 hours in adults and 1 and 48 hours in children. The frequency of attacks is extremely variable, from a few in a lifetime to several times a week, and the average migraineur experiences from one to three headaches a month. The head pain varies greatly in intensity.
The pain of migraine is invariably accompanied by other features. Nausea occurs in almost 90 percent of patients, while vomiting occurs in about one third of patients. Many patients experience sensory hyperexcitability manifested by photophobia, phonophobia, osmophobia and seek a dark and quiet room. Blurred vision, nasal stuffiness, diarrhea, polyuria, pallor or sweating may be noted during the headache phase. There may be localized edema of the scalp or face, scalp tenderness, prominence of a vein or artery in the temple, or stiffness and tenderness of the neck. Impairment of concentration and mood are common. Lightheadedness, rather than true vertigo and a feeling of faintness may occur [1]. We will concentrate our attention on the migraine pathogenesis, taking in consideration involved afferent and effer-ent functions of primary and secondary neurons, with relative neurotransmitters.
GLUTAMATE AND MIGRAINE
Afferent fibres of nociceptive primary neurons are ending in the superficial part of the ipsilateral spinal horn after having contracted synaptic connections with the second order excitatory glutamatergic neurons. Glutamate is able to activate different types of receptors, ionotropic and metabotropic, located centrally as well in the periphery. AMPA and kainate receptors, ionotropic, provoke excitatory post-synaptic potentials (EPSPs) having a rapid onset and a short duration (few ms). NMDA receptors, also ionotropic, induce slower EPSPs, lasting about 100 ms. Metabotropic glutamate receptors, called mGluR, play an important modulating role on the hyperalgesic response; for some of them, like mGlu 4 and mGlu 6-8, positive, for others (mGlu1 and mGlu 5) negative. They activate, through G-proteins, intracellular metabolic chains. The phosphorilation of intracellular substrates like ionic channels is able to modulate different phases of the synaptic transmission and the activation of mGlu 5 can increase the sensitivity of ionotropic receptors also for the glutamate itself. All different glutamate receptors seem to be involved in the phenomenon of hyperalgesia (lowering of nociceptors threshold and intensity), bringing to the elevation of pain induced by noxious stimuli and chronic pain mechanisms. Central sensitization phenomena to which the hyperalgesia is connected seem to depend significantly from the NMDA and mGluR receptors activation operated by glutamate [2].
The spreading depression (SD), characterized by a massive failure of ion homeostasis associated to a transient cessation of neuronal function, and believed to be involved in migraine pathogenesis, requires the release of glutamate; NMDA receptors play a crucial role in the propagation of this process. In isolated layers of murine brain enthorinal cortex slices containing the NMDA–receptor-mediated component of extracellularly recorded field EPSPs, the electrical stimulation performed in order to elicit spontaneous SD , in the presence of ifenprodil, a selective NMDA receptor antagonist, was nearly unable to produce the occurrence of SD [3].
In anaesthetised cats, recordings of the trigemino-cervical complex activity evoked by electrical stimulation of superior sagittal sinus, in cells activated by L-glutamate, showed that CGRP receptor antagonists alpha-CGRP and BIBN4096BS, given by microiontophoresis onto neurons in the trigemimovascular complex or intravenously, significantly inhibit the evoked activity. Data obtained suggest that there are non-presynaptic CGRP receptors in the trigeminocervical complex that can be inhibited by CGRP receptor blockade and therefore that a CGRP receptor antagonist could be effective in the acute treatment of migraine and cluster headache [4]. An in vitro study in human embryonic kidney has shown that a selective and non-competitive GLU-K5 antagonist is able to inhibit l-glutamate and domoate- evoked currents blocking the kainate subtype of glutamatergic receptors [5]. Nociceptive spinal neurons present a high degree of plasticity revealed by the increased intensity and duration of neuronal discharges after repeated injuries and prolonged activation of C fibres. Neuronal plasticity and Long Term Potentiation (LTP) processes could be responsible for chronic pain manifestations which are at the basis of chronic migraine. In the LTP genesis is fundamental the NMDA receptor activation which induces a raise of intracellular Ca++ concentrations stimulating NO-synthase (NOS), a Ca++ - calmodulin – dependent enzyme, able to provoke an increased NO formation. This last compound is active at the level where it is produced as well as in the extracellular space, reached by diffusion, and in this place stimulates the guanilato-cyclase of adjacent neuronal endings, provoking a further release of glutamate.
The repeated and intense stimulation of NMDA receptors occurring during migraine attacks and the consequent onset of sensitization could explain the progressive worsening leading from a form of headache showing many free intervals to the daily inter-critical chronic headache which obliges the patient to daily administrations of analgesics under the risk of abuse.
NMDA antagonists like ketamine, given subcutaneously or intravenously by daily continuous infusions, have been assayed in order to avoid the chronic evolution and the addiction toward analgesics, frequently observed in these conditions [6].
A rational perspective for the future is represented by antagonists and modulators of ionotropic or metabotropic glutamate receptors which could be therapeutically efficacious in the treatment of chronic forms of migraine and other types of primary headache. It has been recently shown that the glutamatergig system is implicated in cortical spreading depression, trigemino-vascular activation and central sensitization in preclinical models of migraine, and in the clinic [7].
OPIOID PEPTIDES AND MIGRAINE
The transmission of nociceptive messages from afferent primary fibres as well as the activity of spinal nociceptive neurons are modulated by presynaptic and postsynaptic mechanisms, mediated through different subtypes of spinal interneurons (gabaergic, glycinergic and enkephalinergic).
The first control station of painful afferences is the gelatinous substance (GS) located in the spinal grey posterior horn. GS is rich of opioid peptides (enkephalins and dynorphins) and, of course, of opioid receptors mu and k as well as of theirs interneurons, which selectively inhibit neurotransmitters release from A-delta and C efferent fibres, so making an access gate for painful afferences. Opioid peptides exert a prevalent inhibiting effect on neuronal activity due to the opening of K+ membrane channels, operated by mu and delta receptors, or to the closing of Ca++ channels (k receptors).
Spinal nociceptive circuits and especially enkephalinergic neurons are under the control of neuronal systems descending from the brainstem able to exert facilitating or inhibiting effects and therefore playing an important role in the regulation of nociceptive reflexes and pain perception.
The grey periaqueductal substance (PAG), the raphe magnum nucleus (NRM), reticular paragigantocellular (NRPG) and magnocellular (NRMC) nuclei form all together the inhibitory descending system. PAG neurons project upon NRM, where arise descending serotoninergic fibres and NRPG as well as NRMC, and from this last structures take origin noradrenergic descending fibres, directed, as the serotoninergic fibres, to the spinal cord. Serotoninergic and noradrenergic pathways activate spinal enkephalinergic interneurons, negatively modulating, through a presynaptic inhibition, the transmission of the nociceptive message. In this way opioid agonists, like morphine, fentanyl, pentazocine , buprenorphine and others,exert their analgesic effect.
Though the opioid system has received not a major consideration in the study of migraine pathophysiology, its involvement can be postulated. As far as morphine is concerned, according to several observations, it seems that this drug could worsen rather than attenuate the migraine pain.
In United States only butorphanol has been employed by intranasal route in the therapy of very severe migraine attacks. It has been observed the absence of interaction between butorphanol and sumatriptan also when the administration of these two drugs is practised closely in time [8].
Recently it has been found that the gene codifying for mu receptors is polymorphous and this peculiarity could explain the individual variability to stimuli generating pain. Since migraine is very likely a disorder characterized by a variable threshold and a multiform pathogenesis, both characters being tied to different genes, it is not possible to exclude that the mu opioid receptor could represent one the factors contributing to the determination of migraine threshold and on this basis it will be rational to address in such direction future studies on migraine genetic aspects, in order to evaluate precisely the eventual involvement of all sub-types of opioid receptors.
Opioid analgesics appear to be generally inefficacious in the management of chronic daily headache. The study of pathophysiological models of opioidergic pathways for migraine and chronic daily headache, as well as for neural plasticity in the context of neuropathic pain states, have allowed the concept of chronic daily headache as a neuropathic pain syndrome [9].
SEROTONINERGIC RECEPTORS AND MIGRAINE
The experimental research in animal models has given in these last years the opportunity to obtain a large amount of significant data on the role of trigeminal receptors in the pathogenesis of migraine and cluster headache. Although the knowledge of complex pathogenetic factors involved at the level of the central nervous system in the production of the migraine attack is still full of gaps, we dispose of a considerable information on peripheral factors presumably responsible of pain mechanisms in migraine and other types of primary headache.
The brain is not provided with pain sensitive endings, while meninges are rich of nociceptors. The ophthalmic branch of trigeminal nerve reaches extra-cerebral vessels at the level of meninges (dura mater, arachnoid and pia mater) which receive peripheral fibres coming from pseudo-unipolar neurons present in the trigeminal ganglion. These fibres form the neuroanatomical circuit known as trigemino-vascular system, forwarding stimuli received from afferent peripheral endings to the trigeminal caudalis nucleus and to other brainstem nuclei as superior salivatorius nucleus. According to Edvinsson and Goadsby [10], through this last nucleus could be activated an afferent cholinergic pathway of the seventh cranial nerve, with the release at the meningeal level of Vasoactive Intestinal Peptide (VIP), able to provoke further vasodilatation and hyperactivation of trigeminal fibres An important role is attributed to parasympathetic pathways in migraine pathogenesis specially in order to explain some symptoms like tearing, conjunctival blood injection and rhinorrhea observed frequently in patients suffering from cluster headache.
The trigemino-vascular system, therefore, with its different components, represents one of the most important anatomic and functional areas for the study of migraine pathophysiology.
The characterization of 5-HT receptors within trigeminal sensory neurons has deeply improved the knowledge about the pathogenesis and treatment of migraine; among them 5-HT1B are located in the smooth muscle of meningeal extracranial vessels, 5-HT1D in trigeminal sensory endings [11] and 5-HT1F at the level of trigeminal nucleus caudalis, as well as peripherally in trigeminal gangliar endings [12]. The stimulation of first two sub-types of 5-HT receptors inhibits the release of vasoactive peptides like CGRP and pain transmitter peptides like substance P and neurokinin A [13]. The peptides are involved in the dural plasma extravasation provoked by the neurogenic inflammation due, in experimental animal models, to the depolarization following the electric stimulation of peripheral trigeminal fibres. In these models the release of CGRP caused by the activation of trigeminal fibres leads to a further dilatation of meningeal vessels, wich, in turn, brings about the hyperactivation of central and peripheral trigeminal endings responsible of pain (Fig.1). These pathophysiological mechanisms,. discovered by Moskowitz [14], could be, in the rat, at the basis of migraine pain, but this hypothesis has been not yet definitely validated in the man, though it has received a strong support from the significant therapeutical activity exerted by triptans toward the primary headache pain. Actually it is well known that sumatriptan inhibits the CGRP gene transcription in trigeminal sensory neurons, but it has not been clarified yet whether this action is mediated only by 5-HT1D or also by 5-HT1B receptors, the last having been isolated, as the relative RNAm, in human trigeminal gangliar neurons, in absence of an histochemical demonstration of this receptor transportation at the level of central and peripheral endings, as it happens for 5-HT1D [12], which is present also at the level of the solitary tract nucleus. In this site, believed to be involved in the pathogenesis of some migraine symptoms like nausea and vomiting for its connections with the autonomous nervous system, arrive numerous trigeminal afferent fibres and, therefore, the therapeutic activity of triptans on the above mentioned symptoms, associated to the analgesic effect, could be exerted either directly on the solitary tract nucleus or indirectly, as a consequence of the reduced activation of the trigeminal nociceptive system.
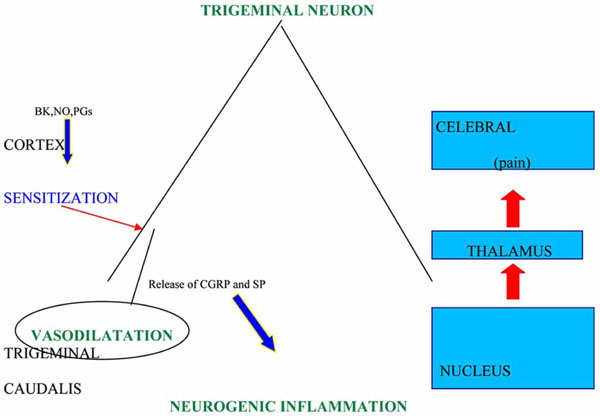
Attack eliciting factors at the sub-cortical level provoke dilatation of cranial vessels with the activation of trigeminal sensory afferent endings and a consequent neurogenic inflammation process, due to the release of CGRP and SP or NK-A.
BK: bradykinin; NO: nitric oxide; CGRP: calcitonin gene-related peptide; SP: substance P; NK-A: neurokinin A.
Another indirect proof of the possible application to humans of the neurogenic inflammation theory of Moskowitz seems to reside in some observations made by Goadsby et al. [15], according to them CGRP levels increase in the external jugular vein blood during migraine attacks.
On the other hand, the same authors showed the correlation existing between the decrease of blood CGRP levels and therapeutic efficacy of triptans. Through their agonist activity on 5-HT1 B-D-F receptors these drugs could inhibit at the presynaptic level the release of neuropeptides able to provoke either intracranial vasodilatation or pain characteristic of migraine and related to the stimulation of primary trigeminal nociceptive neurons, which represent the first step in the pain transmission. These receptors, in turn, activate, through their central endings, second order nociceptive neurons at the level of brainstem, in the trigeminal nucleus caudalis where also 5-HT1 F receptors are located as well as at peripheral endings of gangliar trigeminal neurons in which they could have a function analogous to that of 5-HT1 D receptors [12].
Antagonists of 5-HT1 F like LY 3334370 inhibit selectively, in anesthetized rats, the activation produced by the electrical stimulation of dura mater and of second order neurons in trigeminal nucleus caudalis, in this way blocking the transmission of nociceptive inputs to central areas [16].
Since many triptans show to be provided with a 5-HT1 F agonist activity it is possible to hypothesize that their therapeutic efficacy be partially due to the ability by them possessed to activate this sub-type of 5-HT1 receptors either at peripheral levels or, perhaps more likely, centrally, in correspondence of the trigeminal nucleus caudalis [16,17].
Preclinical investigations in rats have shown that anti-migraine triptan drugs exert their effect not only by blocking the neuropeptide neurotransmitter release from sensory nerve terminals and directly constricting blood vessel smooth muscles, because also the modulation of glutamate and NO release by them operated can contribute to their activity [18,19] . The majority of 5-HT1B, 5-HT1D and 5-HT1F are co-located with glutamate receptors being glutamate positive ,as it is possible to demonstrate using techniques of immunohistochemical staining [20]. According to Levy et al. [21], triptans analgesic action is exerted through presynaptic 5-HT1B-D receptors in the dorsal horn by blocking the synaptic transmission between axon terminals of the trigeminovascular neurons and cell bodies of their central counterparts. On this basis the analgesic action of triptans can be attained specifically in presence and not in the absence of central sensitization. The early treatment with triptan drugs provides a powerful means of preventing the initiation of central sensitization triggered by chemical stimulation of menigeal nociceptors [22]. An important site of action for triptan anti-migraine drugs is the nucleus tractus solitarius where trigeminovascular activation triggers nausea and vomiting, frequently associated with pain in the migraine attack [23].
GABAERGIC RECEPTORS AND MIGRAINE
Among receptors with a prevailing inhibitory activity an important role is played by GABA receptors, in particular in the neurogenic inflammation process, as it has been proven in animal experimental models using valproate [24], an anticonvulsant drug currently used in the preventative treatment of migraine, also assayed as a symptomatic of second order in the therapy of cluster headache [25]. Good results have given in the treatment of primary headache also more recent anticonvulsant like gabapentin, lamotrigine, tiagabine, topiramate and others [26, 27].
GABA-A receptors are located in trigeminal sensory neurons. Valproate is provided with two mechanisms of action, being able to inhibit GABA-transaminase, which causes GABA degradation and, at the same time, to activate glutamic - decarboxylase, that induces GABA synthesis. Therefore, following its administration, GABA extracellular concentrations in the proximity of GABAergic endings are increased [28].
Valproate, in experimental animal models, blocks dural plasmatic extravasation induced by neurogenic inflammation due either to the electric stimulation of trigeminal fibres or to the intravenous administration of substance P. For this aspect preclinical data have been obtained later than clinical evidences, when the use of valproate in migraine therapy was already consolidated, but anyhow they bring a strong support to the hypothesis that the activation of trigeminal sensory neurons is an initial step in the neurogenic inflammation process and in the pathogenesis of primary headache pain.
About GABA-B receptors they have also been implicated with migraine pathogenetic mechanisms. The coupling between GABA and GABA-B receptors induces the activation of a specific Gi-protein, exerting an inhibitory effect on the enzyme adenylate-cyclase with the reduction of intracellular cAMP levels and decreased phosphorylation: Consequently it is produced a functional block of voltage-dependent Ca++ channels involved in the neurotransmitter release. GABA-B agonists, as baclofen, then, may negatively modulate, trough these mechanisms, the release of many neurotransmitters, GABA included. Baclofen indeed has shown to inhibit the activation of trigemino-vascular system at the level of the spinal trigeminal caudatus nucleus and therefore to be furnished of a distinguished anti-nociceptive activity, observed in many clinical trials on patients suffering from migraine or cluster headache [29,30].
CANNABINOID, VANILLOID AND PAR RECEPTORS RELATED DRUGS AND MIGRAINE
At the level of peripheral and central endings of primary sensory neurons are present also cannabinoid, vanilloid and PAR receptors, characterized only few years ago (Table1), which seem to play an important role in pain pathophysiology Endocannabinoids (anandamide and 2-acyl-glycerol , having a lipidic structure, derive, like eicosanoids and Platelet Activating Factor (PAF), from membrane phospholipids and their pharmacodynamic profile is similar to that of the Cannabis active principle delta-9-tetrahydrocannabinol and other natural and synthetic cannabinoids. The synthesis of anandamide is regulated by the phospholipase D, while another intracellular enzyme, the Fatty Acid Amide Hydrolase (FAAH) is responsible of its degradation, followed by the reassumption of its constituents in phospholipids [31]. Differently from neurotransmitters and neuropeptides which are continuously synthesized and then stored in special deposit sites, from where they are released in consequence of the excitation of nerve endings, endogenous cannabinoids are produced from endothelial cells, macrophages and peripheral cells only when their holocrine-paracrine function has to be developed, during the inflammation process and in all conditions of cellular injury [32, 33].
Receptors Present at the Level of Sensory Primary Neurons and Possible Effects on the Release of Neuropeptides Involved in Neurogenic Inflammation and Migraine Pain
| RECEPTORS | LIGANDS | ACTIVITY ON THE SENSORY PRIMARY NEURON |
|---|---|---|
| 5-HT1-D | SEROTONIN | DECREASED RELEASE OF CGRP AND SUBSTANCE P |
| GABA-A | GABA | DECREASED RELEASE OF CGRP AND SUBSTANCE P |
| CB-1 | ANANDAMIDE* | DECREASED RELEASE OF CGRP AND SUBSTANCE P |
| VR-1 | CAPSAICIN OR ANANDAMIDE** | INCREASED RELEASE OF CGRP AND SUBSTANCE P |
| PAR-2 | PEPTIDIC CHAINS SLIGRL AND SLIGKV | INCREASED RELEASE OF CGRP AND SUBSTANCE P |
* At nanomolar concentrations.
** At micromolar concentrations.
Once released in the extracellular place, anandamide activates, at nanomolar concentrations,its own receptors defined cannabinergic (CB) and generally distinguished in CB-1 and CB-2, while, at micromolar concentrations [34], stimulates vanilloid receptors (VR-1).
CB receptors of both types are coupled, through a Gi- protein, with the adenilate cyclase [35]. CB-1s mediate analgesic effects of anandamide either peripherally or centrally; CB-2s, present in lymphocytes B and natural killer, seem able to produce, when activated, an anti-inflammatory action.
Anandamide (AEA) thus exerts centrally and peripherally an inhibitory effect on the pain, due to the activation not only of encephalic and spinal (10th lamina) CB receptors, as in the PAG [36], but also of peripheral sensory neurons [37], in particular at the level of central peptidergic capsaicin-sensitive endings, containing CGRP and substance P, where their release is reduced [38].
Hohmann and Herkenham [39] have demonstrated, by histochemical techniques, the presence of CB-1 also in the peripheral endings. Calignano et al. [40] showed that CB-1 agonists, like WIN552122, when applied on the skin, are able to reduce the nociceptive activity of formalin.
After a protracted and intense noxious stimulation, sufficient to provoke an inflammatory reaction, central and peripheral neuronal excitability is significantly increased until the onset of hyperalgesia and pain production by normally innocuous events (allodynia). Cannabinoids, in animal experimental models, antagonize either hyperalgesia or allodynia [41-43]. On the other hand, CB-1 antagonists, like SR141716A, decrease the activation threshold, increasing the response to mechanic and thermic stimuli.
Therefore, it may happen that, following repeated or protracted noxious stimulations, as after a traumatic injury or in course of inflammation, endocannibanoid levels appear increased either peripherally or centrally, for example in the PAG, while at the same time a sensitization of CB-1 receptors is observed. Thus experimental data seem to demonstrate the central role of endocannibanoids in pain pathways, disclosing the perspective of a possible interaction on these new targets in the treatment of various clinical forms of pain.
Anandamide, therefore, activates, respectively at nanomolar and micromolar concentrations, CBs and VR-1 receptors, these last capsaicin-sensitive [34], with opposite effects on pain, toward which it exerts a dual activity, stimulatory or inhibitory, so assuming in the regulation of this function, a balancing role. Vr-1 receptor, discovered in 1977, when stimulated, provokes the inflow of extracellular Ca++ in response to the loss of intracellular Ca++ [44].
Vanilloid receptor VR-1 is a non selective receptor-channel, which can be activated by protons as well as by capsaicin-like substances with a simil-lipidic structure, producing the release of inflammation mediators with pain; moreover it is able to transduce also heath stimuli in nociceptive.sensations. Capsaicin-sensitive neurons have a small diameter (A-delta and C fibres) and are peptidergic, releasing CGRP and substance P through a Ca++ - dependent mechanism, not only at the peripheral level but also centrally, in spinal endings of primary sensory neurons [45]. Therefore they can exert either peripherally or centrally both the analgesic or algesic effects on nociception, sustained by opposite modifications of Ca++ intracellular content [46]. Ethanol potentiates VR-1 nociceptive response [47].
It is not yet possible to argue whether such a mechanism could or not be involved in the neurogenic inflammation process, validating in the first case Moskowitz’s theory [14].
Another class of receptors, named PAR (Protease Activated Receptors) has been isolated in the rat, by means of immunohistochemical techniques [48], in capsaicin-sensitive primary sensory neurons, at the level of trigeminal and spinal dorsal root ganglia, where they are colocated with VR-1 receptors, being probably involved also in primary headache pathogenetic mechanisms.
The PAR-2 receptor [49] is selectively activated by SLIGRL and SLIGKV peptides, trypsin and tryptase, proteases abundantly contained in mast cells and released by them in course of the inflammatory process; in many tissues mast cells are in close contact with the endings of capsaicin-sensitive primary sensory neurons containing neuropeptides. The activation of PAR-2 provokes Ca++ mobilization and CGRP and substance P release in slices of rat isolated neurons or vascular intracranial structures cultured in vitro, while in vivo the swelling CGRP-induced of the rat paw is observed.
Therefore CB, VR and PAR receptors with their ligands seem worthy of attention in the study of pain pathophysiology and therapeutical problems related to the migraine [50-53].
CONCLUSIONS
The complexity of interactions taking place in the sensory neuronal network with the mediation of all different neurotransmitters involved gives the measure of the extreme difficulty connected with the knowledge of migraine pathogenesis. Many theories have been formulated in these last sixty years about the pathogenesis of migraine and other forms of primary headache, but the problem is still far to be fully clarified.
The first theory, conceived by Wolff in 1948 [54], is build up on a vascular basis, considering three main aspects: a) during the migraine attack extra-cranial vessels dilate and are throbbing in a large percent of patients; b) the stimulation of intra-cranial vessels provokes an ipsilateral headache; c) vasoconstrictor drugs, like ergot derivatives, show a curative effect, while vasodilators, like nitrates, may induce an attack. On this observations he hypothesized that an intra-cranial vasoconstriction could be responsible of the migraine aura and of the following hyperemic reaction associated to local vasodilatation, with consecutive activation of perivascular nociceptive endings. Ten years later Heyck [55] completed the theory adding the concept that during the attack arterio-venous anastomoses were closed, explaining in this way the decreased oxygen extraction observed in the symptomatic side.
Successive theories were underlining the importance of vasoactive and neuroactive substances (as plasmatic kinins, endorphins, serotonin, histamine, fatty acids, adenosine, prostaglandins, NO and endothelin-1), released in perivascular areas, to which could be attributed the responsibility of the neurogenic inflammation and its symptoms [56-59].
The already mentioned theory of Moskowitz [14] was synthesizing in an appreciable way all theories till now exposed, with the idea of the neurogenic inflammation in the trigemino-vascular area and the integration in the cerebral cortex of the painful information.
Another possibility to interpret the migraine pain is proposed on the basis of a depression of the cortical electrical activity similar to the spreading depression observed in animal experimental models by Leao longtime ago [60] and consisting in the production of cortical hyperexcitation waves followed by the electrical activity suppression after chemical or mechanic stimuli, this last progressing by contiguity within the cerebral structures in posterior-anterior direction, at a speed of 2-5 mm/min [61]. The spreading depression can be considered, according to Olesen [62], the pathophysiological substrate of the migraine aura, during which it has been observed with the technique of xenon 133 a decreased cerebral blood perfusion specially in the occipital area, lasting about an hour. These data have been validated by means of magneto-EEG, SPECT, MRI and PET studies [63], specially for the migraine with aura and, at least partially, also for the migraine without aura. Mitochondrial alterations and a decrease in Mg++ blood levels [64] have also been associated to migraine in neuroimaging investigations [65].
Lance [66], Diener and May [67], and Welch [68] have proposed to consider the participation in the migraine pathogenesis of orbital, frontal and limbic cortical areas and brainstem structures specialized in controlling pain mechanisms , like trigeminal nucleus caudalis, locus coeruleus, raphe nuccleus dorsalis,, PAG and area postrema, interacting with several neurotransmitters as noradrenaline, serotonin, dopamine and others, which could be “migraine generators”.
About the cluster headache, among other theories, it seems worthy of interest the immunological theory, dealing mostly with interleukins and cytokines receptors and involving the complex relationships between neuro-psychic, endocrine and immunological systems [69], as it is demonstrated by the reduced response of cortisol to meta-chlorophenyl-piperazine [70]. During the attack there is the activation of the hypothalamic grey ipsilateral to the side affected [71].
As far as tension migraine episodic and chronic, is concerned, it is not clear at all whether its pathogenesis is similar or different, and in what extent, in respect to that of migraine, but many resemblances have been recognised. The influence of stress and of protracted muscular contraction has been largely investigated as well as immunological implications, specially in the autoimmune sense [72].
It is necessary to assess the importance of genetic factors in the pathogenesis of headache and headache pain [73].
Finally, it can not be omitted that the knowledge of migraine pathophysiology has significantly improved because of the advances realised by clinical pharmacology studies, from which further and ever more important insights in the field likely are going to be reached in a next future.

