All published articles of this journal are available on ScienceDirect.

Nrf2-Mediated Signaling as a Therapeutic Target in Alzheimer’s Disease
Abstract
Nrf2 is a major transcriptional factor that controls gene expression in normal health and pathological conditions. It regulates and controls the manifestation of various major elements of oxidative stress, neuro-inflammation, autophagy, and mitochondrial bioenergetics in the centre and periphery. Besides, Nrf2 activity is also controlled at various stages, such as protein degradation, transcription, and post-translation. Growing evidence suggests changes in the levels of Nrf2 in degenerative disorders, such as Alzheimer's disease (AD). AD is characterised by elevated oxidative stress, neuro-inflammation, synaptic dysfunction, and proteinopathies, which lead to the progressive loss of memory. A decrease in the expression of Nrf2 and its downstream target genes was identified in AD. Recent studies have shown that Nrf2 interferes with various main pathogenic processes in AD, including amyloid and tau pathologies. The current review focuses on brief in the regulation of Nrf2 and the association of Nrf2 with AD, along with the currently available Nrf2 activators.
1. INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurode- generative disorder that displays amyloid [1] and tau proteinopathies [2]. Apart from these proteinopathies, the classic theme in various diseases, including AD, is oxidative stress. The disparity between the generation of free radicals and the body's natural antioxidant defense mechanism is known as oxidative stress [3, 4]. In mitochondria, peroxisomes, and the endoplasmic reti- culum, Reactive Oxygen Species (ROS) are essential for various endogenous metabolic reactions and molecular signaling pathways [3, 5], and the excess production of ROS damages biological elements, such as DNA, RNA, and proteins [2, 6, 7].
The unregulated redox balance is the collective result of a variety of biological processes, including metal homeostasis and endogenous and exogenous stress [3, 4]. CNS is more prone to oxidative stress due to its high metabolic rate and high ascorbate levels and transition metals, which act as pro-oxidants [8, 9]. Depletion of antioxidants increases the vulnerability to lipid peroxidation, genotoxicity, protein oxidation, DNA and RNA oxidation, and mitochondrial depolarization. Oxidative stress is also produced by excitotoxicity and plasticity [8, 9], which leads to the activation of various transcription factors [10]. The antioxidant system in the body generates various enzymes that scavenge these free radicals and offer cytoprotection. One such defence system is the Nrf2 signaling system [11, 12].
2. STRUCTURE AND FUNCTION OF THE Nrf2
Nrf2 (Nuclear Factor-Erythroid Factor 2) is a 66 KDa protein molecule. Based on molecular architecture, the Nrf2 is categorised into 7 conserved Neh (Nrf2-ECH) domains, namely Neh1-Neh7 [13]. Neh1 region consists of a regular leucine zipper motif, and the Neh2 domain interrelates with Keap1 via ETGE and DLG motifs [14]. This Neh2 domain is required for the stability of Nrf2. While Neh3, Neh4, and Neh5 are essential for the transcription of Nrf2 target genes [15]. Furthermore, Neh6 allows Keap1-independent regulation, caused by GSK3β phosphorylation [16] and the Neh7 domain, associated with the retinoic X receptor a, which aids in the repression of gene transcription of Nrf2 [17].
Nrf2 acts as a master regulator of antioxidant response via increasing antioxidant enzymes, metabolism of xenobiotics, elimination of damaged proteins, suppression of inflammation and metabolic damage, and influencing various growth regulatory factors [18].
3. REGULATION OF Nrf2
There are three major regulatory pathways, where Nrf2 gets degraded via ubiquitin. The first important one is the use of Keap1; second, the use of glycogen synthase (GSK3); and lastly, the β-TrCP-dependent cul-1-dependent ligase complex [18].
3.1. Keap-1 Dependent Regulation
Kelch, like ECH-associated protein 1 (Keap 1), is known to be the new Nrf2-binding protein. It consists of three main domains that interact with the proteins: the BTB domain in the N-terminal region, the IV region in the centre, and Kelch repeats in the C terminal. BTB mediates binding to Cul3 of Nrf2 ubiquitin ligase [19]. The activation of Keap 1 is required for homeostasis, and the depletion of Keap 1 induces cell death [18]. Under physiological conditions, the activity of Nrf2 is inhibited in the cytoplasm by sequestration and degradation of Nrf2 via Keap1. The Nrf2 Neh2 region binds to Keap1 at the area between the BTB and Kelch repeat domains [19]. This interaction acts as an adaptor for the Cul3/Rbx1 E3 ubiquitin ligase complex, which causes the ubiquitination of Nrf2 [20, 21]. Moreover, Keap1 sequesters Nrf2 to the mitochondria via a ternary complex along with the PGAM5 protein [22, 23]. This sequestration is known to increase the activation of Nrf2 in response to stressors [22].
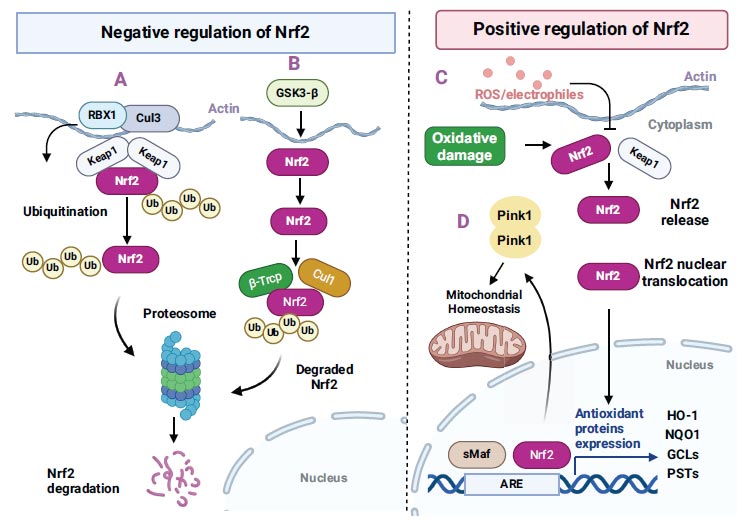
Schematic illustration of Nrf2-mediated regulation in neuronal cells (negative and positive regulation). The image is adapted from Tejo and Quintanilla, 2021. CC BY 4.0 [https://creativecommons.org/licenses/by/4.0/].
3.2. Regulation of Nrf2 via GSK-3
GSK-3β regulates a number of targets in many cell signalling pathways, and it is not astonishing that this enzyme is also involved in the control of Nrf-2 (Fig. 1). It is a constitutively active kinase whose action is nullified by insulin [24]. Increased insulin signaling triggers protein kinase B/Akt through phosphorylation. Consecutively, Akt inhibits GSK-3β via phosphorylation of Ser9. This kinase exerts negative regulation on Nrf-2 by modulating its distribution. Increased oxidative stress conditions cause down-regulation of Akt, stimulation of GSK-3β, and translocation of Nrf-2 from the nucleus to cytoplasm, thus, restricting the antioxidant response [25, 26]. The GSK-3β inhibition results in the elevation of transcriptional activity of Nrf-2 because of nuclear accumulation, thus signifying the fundamental role of the GSK-3β in Nrf-2-ARE down-regulation after oxidative injury [22, 25]. The main mechanism by which GSK-3β mediates Nrf-2 is known to comprise Fyn, a tyrosine kinase that is phosphorylated by activated GSK-3β and causes the nuclear localisation of Fyn. This activated Fyn further causes phosphorylation of tyrosine 568 in nuclear Nrf-2, triggering the inhibition of Nrf-2 protective gene transcription [26, 27].
3.3. Keap 1 Independent Regulation via β-TrCP
Apart from the β-TrCP, the PI3K/Akt pathway also plays a role in Nrf2 induction, independent of KEAP1 [28]. The PI3K/Akt signaling stimulates Nrf2 through the GSk-3β inhibition, which causes phosphorylation of Nrf2, thus causing β-TrCP to consecutively ubiquitinate Nrf2 for consequent proteasomal degradation [25, 29]. The β-TrCP serves as a substratum receptor inside the SCF-ubiquitin ligase β-TrCP and takes part in the ubiquitination of Nrf2 [30]. β-TrCP recognises its substrates by directly attaching to phosphorylated destructed motifs, and this pattern represents the GSK-3β target elements; thus, a variety of its target proteins are known to be processed by β-TrCP.
4. Nrf2 AND ITS ROLE IN AD
AD is a neurodegenerative disorder predominantly exemplified by cognitive dysfunction. Pathologically, the disease exhibits β-amyloid plaques [31] and neurofibrillary tangles [32] as pathological hallmarks. Besides, it also causes oxidative stress [33, 34], neuroinflammation [35, 36], microglial activation [36], astrogliosis [37], and synaptic loss [33, 38]. The amyloid-β proteins of the amyloid plaques and the neurofibrillary tangles disrupt homeostasis and lead to neuronal death and neurodegeneration. The prevalence of AD is known to increase logarithmically with age. The amyloid pathogenesis and tauopathies alter the mitochondrial biogenesis and bioenergetics, which eventually lead to oxidative stress [18]. Post-mortem AD brain studies revealed the presence of high levels of lipid oxidation products, oxidised DNA bases, and carbonyls in proteins [39], which suggests the presence of oxidation of macromolecules in AD brains.
Nrf2 is uniformly found in a different range of tissues and cells, with varying amounts of protein present in particular cell types that are primarily in charge of homeostatic adjustments. Astrocytes and microglia, for instance, play a particularly important role in Nrf2 in the brain since their expression levels are higher than those of neurons [40]. Astrocytes are highly responsive to Nrf2 activation, while the Nrf2 modulates microglial dynamics, thereby reducing the inflammatory mediators and increasing the anti-inflammatory markers [41].
Nrf2 signaling has been widely studied in AD animal models over the last few decades, indicating its downregulation. Nrf2 serves as a main mediator in protecting against oxidative insults. Previous research has shown that the Nrf2/ARE pathway was modulated in AD [34], and the levels of Nrf2 were decreased in the human AD hippocampus [12]. The absence of Keap 1 activated Nrf2 and induced enzymes in phase II reactions in neuronal cells, thereby amplifying resistance to oxidative insults [42]. It has been observed that Nrf2 can hinder AD, and the agents that stimulate Nrf2 have shown neuroprotective activities in preclinical models [43], while the deficiency of Nrf2 exacerbates memory deficits [44]. Furthermore, in the rat model of AD, the Nrf2/ARE levels were found to be downregulated, and the oxidative stress markers, such as the SOD and catalase levels, were decreased. Also, the MDA levels were increased in the cortex and hippocampus [34, 35].
In another study, GSK-3β inhibition in mice was known to raise nuclear Nrf2 in the cortex [45]. Glial activation was found in AD, leading to elevated inflammatory markers, such as IL-1β, TNF-α, and COX2 [34]. Besides, NF-κB was found to be associated near the amyloid plaques. It was also found that the discrepancy between Nrf2 and NF-κB could be a key part of AD pathology. While the NF-κB activation causes the production of pro-inflammatory markers like various cytokines and chemokines that further increase the amyloid-β production and microglia activation [46, 47], NF-κB is associated with β-secretase enzyme expression, as this enzyme possesses a binding site with NF-κB, thus, associating with the inflammatory response [40]. In another study, the molecular association between Nrf2 and inflammation was studied. It was found that Nrf2 restricted lipopoly- saccharide-induced transcriptional stimulation of proinflammatory cytokines like IL-6 and IL-1β. It was also demonstrated that Nrf2-mediated suppression is independent of oxidative stress and Nrf2-binding pattern. Contrary to the widely accepted view, these studies reported that Nrf2 is the upstream controller of cytokines and the basis for the Nrf2-mediated anti-inflammatory approach [48].
4.1. Targeting Nrf2 for AD Pathology
Multi-drug targeted therapy in AD came into existence after the failure of many clinical trials for AD. Target-specific options can be achieved by targeting multiple pathological pathways. One such pathway is the Nrf2 signaling pathway. As discussed above, Nrf2 can significantly offer neuroprotection in AD. So, treatment with Nrf2 activating agents can mitigate AD. Various Nrf2 agents/compounds are listed in Table 1.
| S.No. | Compound/molecule | Models | Uses | Reference |
|---|---|---|---|---|
| 1. | Benincasa hispida | Rat model of AD | Antioxidant, anti-inflammatory | [34] |
| 2. | Carnosic acid | Transgenic mice model | Antioxidant, anti-microbial, anti-inflammatory | [49] |
| 3. | Astaxanthin | 3xTg mice model | Antioxidant, blood pressure regulation, Anti-hyperlipidaemic |
[50] |
| 4. | DMF | Transgenic model | Anti-inflammatory | [41] |
| 5. | Lycopene | Transgenic mice model | Antioxidant, anti-apoptotic | [51] |
| 6. | Phenols (resveratrol, curcumin, gastrodin) | Clinical trials and Rodent models | Antioxidant, anti-inflammatory | NCT00678431, NCT00164749 [52]. |
| 7. | Ebselen | 3xTg mice | Antioxidant, suppression of mitogen-activated kinases | [53] |
| 8. | Tideglusib | Clinical trial | Antioxidant, anti-inflammatory |
NCT01350362 |
| 9. | Flavonoids (Fisetin, Hesperidin anthocyanins, Apigenin) | Mice models | Antioxidant, anti-inflammatory |
[54-57] . |
| 10. | Sulforaphane | Clinical trial | Antioxidant, anti-inflammatory, and anti-apoptotic properties |
NCT04213391 |
CONCLUSION
In this review, we discussed various studies that highlighted the neuroprotective role of the Nrf2 pathway in AD. Nrf2 is a conventional example of targeting oxidative stress, neuro-inflammation, and associated underlying disease pathology. Furthermore, Nrf2 is crucial to redox balance and the control of inflammation, and an enhancement in the effect of Nrf2 could be a possible therapeutic approach for AD. Evidence from animal models revealed that a pharmacological therapy to slow the progression of AD might be possible through Nrf2 activators. Nrf2 activating compounds/molecules are currently approved by the FDA for various neurodegenerative conditions, and some molecules, such as resveratrol, curcumin, and tideglusib (Table 1), are under clinical trials. In summary, the upregulation of Nrf2 leads to the synthesis of antioxidant and anti-inflammatory elements, which improves AD pathogenesis.
AUTHORS’ CONTRIBUTION
All authors have accepted responsibility for the manuscript's content and consented to its submission. They have meticulously reviewed all results and unanimously approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| COX-2 | = Cyclooxygenase-2 |
| IL-1 | = Interleukin 1 |
| NF-kB | = Nuclear factor kappa-light-chain- enhancer of activated B cells |
| PI3K/AKT | = Phosphatidylinositol 3-kinase/protein kinase B |
| TNF | = Tumor Necrosis Factor |

