All published articles of this journal are available on ScienceDirect.

Modulatory Action of Voltage-gated Ion Channels in Inflammation and Inflammatory Pain
Abstract
Pain is “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described by the patient in terms of such damage”. The origin of every pain syndrome is inflammation. A group of voltage-gated channels that are permeable to calcium ions enhances sensory transduction witnessed during inflammation. Hence, understanding calcium signaling is an essential step towards recognizing neural network activity associated with pain management. In this review, we attempted to understand the impact of calcium-permeable ion channels in the recognition, processing, transduction and modulation of pain signals. Results obtained revealed that calcium being one of the most ubiquitous secondary messengers plays a significant role in modulating numerous biological processes, including inflammation and pain. Though almost all subtypes of calcium channels are highly expressed in the central nervous system (CNS), the “N-type calcium ion-channels” play an important function at the time of neurotransmitter release from the afferent terminals within the spinal dorsal. Hence, they serve as a key therapeutic target during the treatment of analgesics. Migraine is also reported to involve neurogenic inflammation. “P/Q-type calcium channels” is suggested to have an important role in migraine. The inhibition of these channels through various analgesics serves as a treatment against inflammatory and neuropathic pain. However, few of these inhibitors have numerous side effects, including cancer. Hence, these inhibitors may be consumed under the supervision of medical practitioners. In this review, we revealed the understanding and regulation of ion channels in inflammation causing pain and its treatment.
1. INTRODUCTION
Pain is a debilitating symptom of many diseases and causes significant disability and reduces the quality of life. We still do not have any effective treatment options and it remains major unmet global public health challenge. The difficulty in understanding and managing pain has also led to widespread overuse of, and addiction to opoids/morphine medications. This can lead to serious adverse outcomes [1, 2]. Pain can be classified into three major groups: Nonciceptive, Neuropathic and Nociplastic. Nociceptive pain results from activity in neural pathways and is the most common form of chronic pain encompassing arthritis and most forms of spinal pain. Neuropathic pain is caused by damage or disease affecting the somatosensory nervous system, typically associated with sensory abnormalities such as numbness and alloydynia. Approximately 15-25% of chronic pains are neuropathic in nature and include diabetic neuropathy, postherpetic neuralgia and radiculopathy. Nociplastic pain is the pain arising from the abnormal processing of the pain signals without any clear association with damage, injury or disease pathology [3, 4]. Pain may be either acute or chronic [5]. Acute pain is an essential sensory signal that protects us from harmful environmental stimuli, for instance, cold, heat, tissue damage or chemical irritants. Without acute pain, it would be hard to protect against harmful behavior [6]. On the contrary, chronic pain is “pain which has persisted beyond normal tissue healing time” [7]. Chronic pain is one of the common problems associated with anxiety, depression, opioids dependence, and restrictions in mobility and daily activities [8]. Ruan et al., gave evidence that the overall burden of disease of chronic pain along with its associated comorbidities is unparalleled at the global level [9]. Recently, Mills and the team reported that ~1.9 billion people are affected via recurrent “tension-type” headaches globally, which are the most common symptom of chronic pain [10]. Another systematic review revealed that chronic pain distresses affects ~50% of the total UK population [11]. Across Asia, the incidence of chronic pain in adults ranges from 7% (Malaysia) to 60% (North Iraq and Cambodia) [12]. Irrespective of the high incident rate, chronic pain remains ineffectively treated globally. If not treated early, chronic pain may affect individual emotional as well as physical conditions, thereby declining their work productivity as well as their quality of life. This, in turn, imposes a more significant threat to the socio-economic situation of the patients and their associated people [12].
Most of the studies performed to date suggest that the origin of every pain syndrome is inflammation and inflammatory responses [13]. Modification in gene expression, the voltage dependence of voltage-gated Na+, Ca2+ and K+ channels and ligand-gated ion channels, for instance, purinergic receptors and TR (transient receptor potential) channels, is reported to contribute to the enhanced sensory transduction witnessed during inflammation [14]. A group of these channels that are permeable to calcium ions mediates several signaling functions, for instance, neurotransmitters release, calcium-dependent enzyme activation and calcium-dependent gene expression modification. Hence, understanding calcium signaling is an essential step towards recognizing neural network activity associated with pain management [6]. Thus, in the present review, we attempted to understand the impact of calcium-permeable ion channels in the recognition, processing, transduction and modulation of pain signals. In the near future, this information will be helpful in pain management.
2. NEUROLOGY OF PAIN
To understand how calcium channels are associated with different pain states, it is highly essential to understand the overall facets of the transmission as well as the regulation of pain signals throughout the nervous system. In recent years, several studies have been performed to unravel the mechanism via which different stimuli activate the pain sensors (nociceptors). One of the most critical findings is the identification of “Transient Receptor Potential” (TRP) channels. This ion channel family is highly conserved throughout evolution [15]. It is present in the cell membranes of a broad-spectrum of species, including invertebrate, vertebrate and fungi and is associated with several functions. Few members of this channel family that are situated within the mammal’s nociceptive nerve endings are initiated via thermal stimuli [16]. For instance, heat above 43°C stimulates the TRP vanilloid 1 (TRPV1) channel, which in turn causes an inflow of positive (Na+ and Ca2+) ions; thereby causing cell membrane depolarization. On the contrary, the Transient receptor potential Melastatin 8 channel (TRPM8) receptor retorts to both menthol as well as cold temperatures. This might be the main reason for feeling cold sensation after menthol ingestion. Additionally, numerous receptors are situated at the nerve ending and are activated via various biochemical compounds, e.g., bradykinin and prostaglandins, in response to tissue damage as well as inflammation. Few of these biochemical compounds can even directly modify neural excitability via stimulating ions channels present on the cell surfaces, e.g., ATP and serotonin. Other few compounds, for instance, nerve growth factor (NGF), modulate their effect via second messengers [17].
Transduction, the release of transmitter and transmission of the action potential are the three major physiological processes required for the peripheral transmission of sensory stimuli. Specialized ion channels and receptors (such as transient receptor potential vanilloid 1 (TRPV 1)) are present in the peripheral ends of nociceptive neurons and are the potential in transducing painful stimuli into an electrical signal. The amplification of these impulses is facilitated by voltage-gated sodium channels (NavS) till the threshold for action potential generation is met. The transmission of action potentials to the central terminals of the spinal cord is aided by voltage gated-potassium channels (VGKCs) and Navs [18]. Nav1.1, Nav1.6, Nav1.7, Nav1.8, and Nav1.9 are the most prominent Navs found in sensory neurons [19]. Nav1.1 and Nav1.6 are primarily expressed by myelinated A-fiber neurons, whereas Nav1.7, Nav1.8, and Nav1.9 are primarily expressed by unmyelinated C fibers.
TRPM8 is a calcium-permeable, nonselective cation channel. The TRPM8 channel is significant among the thermal-sensing TRP channels for sensing cold temperatures (8–26°C) and adding to the cooling sensation through compounds like menthol and icilin. TRPM8 is expressed on a δ- and C- sensory nerve fibers, as well as dorsal root ganglion (DRG) and trigeminal ganglia (TG) neurons [20]. TRPM8 activation on macrophages has been shown to cause an anti-inflammatory response, with enhanced interleukin 10 (IL-10) and reduced tumor necrosis factor release. TRPM8 activation on pulmonary epithelial cells, on the other hand, promotes the expression of pro-inflammatory cytokines such as interleukin 1 (IL-1) and tumor necrosing factor (TNF) [21].
Out of all biochemical compounds, glutamate is the key transmitter released via the primary afferent neuron terminals. Two forms of glutamate receptors are required for modulating these signals. They function as cation channels within the spinal cord dorsal horn and activate second-order neurons. Α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors are involved primarily in the conduction of day-to-day physiologic pain. N-Methyl-D-aspartic acid or N-Methyl-D-aspartate (NMDA) receptors are distinctive in that they act as both voltages gated and ligand gated. These receptors play a special role in the development of chronic pain and -wind-up states [22].
For initiating short term pain, glutamate initiates AMPA-receptors on neurons present in the spinal trigeminal complex and dorsal horn. In prolonged pain, glutamate initiates the NMDA-receptors that are mainly associated with sensitization. A few other critical biochemical compounds, namely, substance P, ATP and calcitonin gene-related peptide, are also released from the primary afferent terminals. These compounds are mainly associated with sensitization as well as pain prolongation [15]. However, it is still unclear if the receptors responsible for modulating the function of both peripheral as well as central nerve terminals of the primary afferents are the same. For instance, bradykinin-, ATP- prostaglandin-receptors and calcium & TRPA1 channels are responsible for enhancing the release of the transmitter (e.g. Glutamate) and, hence, increasing the pain intensity. Additionally, presynaptic opioids, glycine, cannabinoid and γ-amino butyric acid (GABA) receptors are associated with reduced transmitter release from the central nociceptive nerve terminals, thereby causing less pain [15].
Another significant pathway that is associated with the pain matrix is the “descending pain modulatory system” with noradrenaline- and serotonin-comprising projections from the locus coeruleus and raphe nuclei, respectively. During normal scenarios, initiation of these pathways impedes pain signaling either by inhibitory interneurons present in the spinal trigeminal nucleus and dorsal horn or via direct effects. However, in chronic pain, the balance within the descending pathways as well as the pain matrix gets misbalanced, plausibly via pain itself, or through numerous precipitating factors. Under these circumstances there might also be an alteration of receptor subtypes; thereby making opioids and serotonin for functioning as proalgesic substances [23].
3. CALCIUM ION CHANNEL
Calcium, being one of the most ubiquitous secondary messengers, play a significant role in modulating numerous biological process, including cell proliferation, muscle contraction gene transcription [24]. Calcium ions (Ca2+) play a vital role in cellular organelle function and homeostasis and act as secondary messengers in a variety of cellular activities [25]. Broadly, Ca2+ channels can be categorized into “high-voltage-activated “and “low-voltage-activated” channels. Further, “high-voltage-activated” channels can be subcategorized as L, P/Q, N, R-types. “Low-voltage-activated” channels are also known as T (“transient”)-type [26].
3.1. L-type Calcium Channels
The “L (long-lasting)-type calcium channels are expressed in various cells, including, ventricular skeletal muscle, myocytes, dendritic cells and smooth muscle. L-type calcium channels (LTCCs), also known as Cav1, are voltage-gated calcium channels (VGCCs/Cavs) that are involved in a variety of physiological processes such as muscle contraction, cardiac action potential, neurotransmission, cell cycle, and gene expression, these channels serve as one of the key therapeutic targets during the drug development process [26].
“L-type calcium channels” are comprised of four members, namely, CaV1.1-CaV1.4. Transcripts for all “L-type calcium channels” isoforms are present in the lymphocytes and are mainly associated with endocrine functions [27, 28]. CaV1.1 is mostly present in the skeletal muscle. It is co-expressed along with ryanodine receptors within the GABAergic neurons and produces GABA. They play a significant role in muscle contraction [29]. CaV1.2 and CaV1.3 are mostly present in the shaft & spine of neurons’ dendrites and on the cell soma [30]. They are highly expressed in broad-spectrum of tissues, including cardiac, adrenal chromaffin and neuronal cells and also play a significant job in cardiac pacemaker activity [31]. In cardiac cells, the transmembrane γ-subunit is not present in both CaV1.2 as well as CaV1.3 [31]. In cardiomyocytes, CaV1.2 is associated with the coupling of excitation-contraction. CaV1.3 is also located in the kidney, pancreas and cochlea. In the kidney and pancreas, it modulates endocrine secretion while in the cochlea it modulates auditory transduction. CaV1.4 is mainly expressed within the retinal cells and modulates normal visual functions [32].
3.2. P/Q-type Calcium Channels
Mutations in the α1A subunit of P/Q calcium channel gene CACNA1A encoding the transmembrane pore-forming subunit of CaV2.1 voltage-dependent calcium channel have been linked to a wide spectrum of epilepsy like febrile seizures, early infantile epilepsy and idiopathic genetic epilepsy. CACNA1A mutations have been correlated to familiar hemiplegic migraine, hemiconvulsion-hemiplegia epilepsy syndrome, episodic ataxia type 2, spinocerebellar ataxia type 6 and episodic ataxia type 2 [33].
On the contrary, reduced P/Q channel activity may reduce the risk of ataxia and epilepsy [34, 35]. Amyloid-β (Aβ) oligomers are reported to directly upsurge the recombinant “P/Q-type calcium” current, which in turn may cause neurodegeneration in Alzheimer's disease. However, no specific drug is available for such disorders. As “P/Q-type calcium channels” are mostly expressed in the neurons of the CNS, they serve as an important therapeutic target for drug designing in neurological disorders [36].
3.3. N-type Calcium Channels
N-type calcium channels (also known as CaV2.2) play a significant role in neuropathic pain as well as spinal nociception signaling modulated through Gβγ G-protein subunits. Additionally, it also plays a significant function in the synaptic transmission within the hippocampus. Along with the “P/Q-type calcium channels”, these channels are adequate for synaptic transmission nearby the hippocampal CA3-CA1 synapse. The core unit of the “N-type calcium channels “is formed via the α1B subunit [37, 38]. The synprint region, i.e., intracellular domain amongst the II–III loops of the pore-forming α1 subunit, binds two key constituents of the SNARE complex, namely, synaptotagmin and syntaxin [39]. This subsequently causes calcium influx, which in turn activates vesicle fusion as well as exocytosis through the ‘zippering’ of the plasma membrane with the SNARE proteins [40]. The synprint region also augments with the active zone protein, namely RIM1, which in turn binds with the β auxiliary subunit of “P/Q-type calcium channels” for suppressing inactivation, thereby allowing calcium influx to assist synaptic vesicle docking along with the active zone [41]. Additionally, RIM1 directly ties with the C-terminal area of the α1 subunit of both P/Q- and “N-type calcium channels”, which in turn binds these channels with presynaptic terminals and helps in the release of the synchronous transmitter [42].
The intermolecular interaction between the SNARE complex proteins and “N-type calcium channels” are highly significant because the calcium channel’s II–III loop gets phosphorylated via various kinases, for instance, protein kinase C and calcium calmodulin-dependent kinase II, which in the turn affects the interaction of the “N-type calcium channels” with numerous SNARE complex components and effects the release of neurotransmitter [43]. Nevertheless, it remains elusive to date if any other kinase has an important role in modulating the “N-type calcium channels” function. In 2007, Samuels and the team reported that scaffolding molecule calcium/calmodulin-dependent serine protein kinase (CASK), which is comprised of the binding domain for “N-type calcium channels”, is a cyclin-dependent kinase 5 (Cdk5) substrate [44]. After phosphorylation via Cdk5, CASK upsurges its intermolecular interaction with “N-type calcium channels” for regulating synaptogenesis [45].
Cdk5 is highly expressed in the post-mitotic cells of the CNS and functions after binding with p35. Earlier studies have suggested that Cdk5 is associated with the phosphorylation of numerous substrates; thereby supporting its function in numerous biological processes, including, synaptic plasticity, cytoskeletal dynamics, migration and synaptic vesicle cycle. In excitotoxic environments, an influx of calcium via the NMDA receptors instigates the calcium-dependent protease calpain for cleaving p35 into p25, which subsequently causes hyperactivation of Cdk5 [45]. Earlier studies have also reported the involvement of the Cdk5/p25 complex in various neurodegenerative diseases, for instance, Alzheimer’s disease [45]. Several other studies have also reported the involvement of Cdk5 in modulating synaptic scaling and synapse formation [45] Further, it is also reported that though Cdk5 serves as the key modulator of neurotransmitter release [46], still it is unclear how they modulate neurotransmitter release. Antibodies against the N-type voltage-gated calcium channel (NVGCC) are the second most common antibody found in individuals with Lambert Eaton myasthenic syndrome (LEMS). They are seen in 33–73 percent of patients with paraneoplastic LEMS [47].
3.4. R-type Calcium Channels
The structure of so-called pharmacoresistant (R-type) voltage-gated Ca2+ channels is only partially known. The majority of them are encoded by the CACNA1E gene and expressed as the ion-conducting component as distinct Cav2.3 splice variants (variant Cav2.3a through Cav2.3e or f). The CACNA1E gene has yet to be linked to a hereditary disease, however, it has recently been linked to spontaneous mutations that result in early death [48].
As the “R-type calcium channels” is a crucial source of dendritic Ca2+ influx during action potentials, it is one of the important factors in modulating neural functions [49]. “R-type calcium channels” is associated with broad-spectrum biological processes including myelinogenesis, the physiology of fear, presynaptic/postsynaptic plasticity, pain behavior and neurotransmitter release [50]. It is pertinent to note that “R-type Calcium channels” is also associated with the semaphorin 3A modulated axons conversion into dendrites [51], regulation of neuronal activity at the time of nervous system development [51], vasospasms after subarachnoid hemorrhage in humans [52], protection during ischemic neuronal injury [53] and modulating analgesic opioid effects as well as pain pathways [54]. Even though it remains elusive if single-nucleotide polymorphisms (SNPs) in human CACNA1E gene encoding Cav2.3 have analgesic effects of opioids, recently few studies have claimed an association between fentanyl sensitivity and SNPs in CACNA1E [54]. Expression pattern of “R-type calcium channels” in small to medium muscle afferent neurons is found to be “Cav2.2 > Cav2.1 ≥ Cav2.3 > Cav1.2” [55]. Synaptic levels of R-type voltage-gated calcium channels (VGCCs) are selectively regulated by neuronal activity via synaptic NMDA-type glutamate receptors (NMDAR) signaling and protein translation [26].
3.5. T-type Calcium Channles
Cav3.2 T-type calcium channels are essential pain signal regulators in the afferent pain pathway, and their activity is dysregulated in numerous chronic pain conditions. As a result, it is plausible to assume that blocking T-type calcium channels in dorsal root ganglion neurons and the spinal dorsal horn may be used to alleviate pain. Early pharmacological investigations with T-type channel blockers, such as ethosuximide, and analgesic effects of Cav3.2 channel siRNA knockdown back this up [56].
“T-type calcium channels” work at negative potentials and are characterized via faster and stronger voltage-dependent kinetics of inactivation/activation, quick recovery from inactivation, slow-closing, and reduced single-channel conductance. Their unique overlapping inactivation/activation and hyperpolarization properties permit few calcium entrances even at resting membrane potentials. These distinct properties enable “T-type calcium channels” to function as pacemakers, which in turn helps to prompt sodium-dependent action potentials subsequent to membrane hyperpolarization. Additionally, their oscillatory, as well as rebound burst-firing behaviors, help in modulating normal physiological functions, for instance, thalamocortical-modulated deep sleep. Alike the “L-type calcium channels”, the “T-type calcium channels” show diverse antagonist resistance as well as expression properties. Nevertheless, unlike “L-type calcium channels”, they are stimulated via small single-channel [26]. The “T-type calcium channels” are highly important for post-inhibitory rebound within thalamocortical neurons and within the hippocampal pyramidal neurons in temporal lobe epilepsy [57]. As“T-type calcium channels” are also associated with producing resting inward current, they participate in calcium-dependent ion channels gating and modulation of calcium-dependent expression of genes and enzymes [58]. Mutations in genes encoding the “T-type calcium channels” cause channelopathies, which are clinically characterized via the abnormal biophysical properties as well as cell surface trafficking of the channel, which in turn may cause gain/loss of channel function [59].“T-type calcium channel” dysfunction is also associated with chronic pain and cardiac hypertrophy [37].
4. CALCIUM CHANNELS IN INFLAMMATION AND INFLAMMATORY PAIN
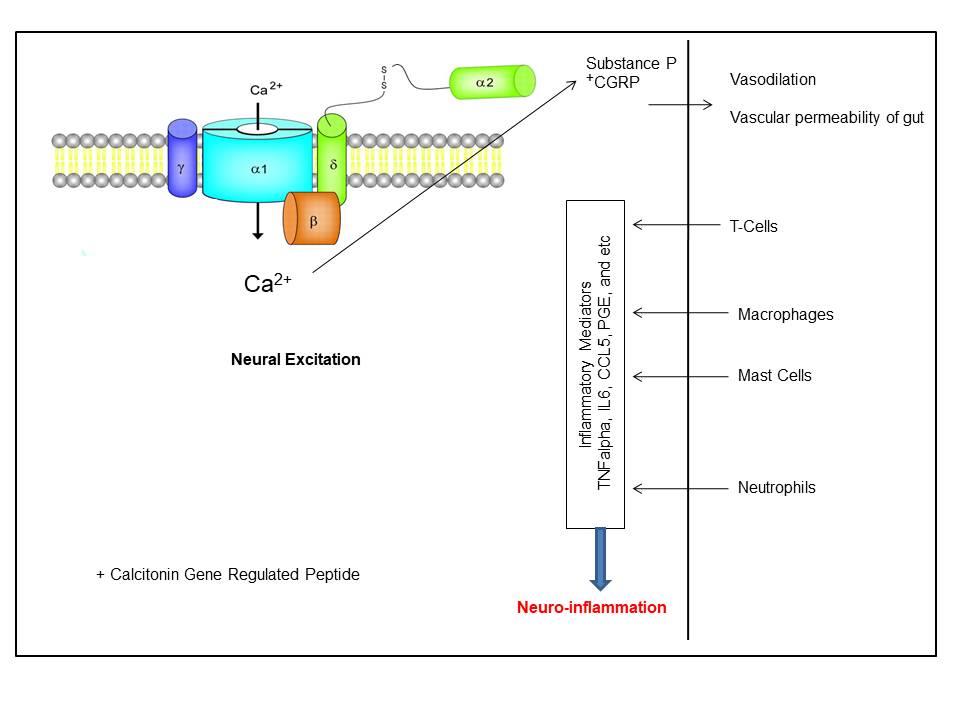
High voltage-activated calcium channels cause extreme primary afferents excitation, which in turn causes neurogenic inflammation and releases neuropeptides, e.g., substance P, from their peripheral endings. This, in turn, stimulates events associated with inflammation, including vasodilatation as well as enhanced vascular permeability (Fig. 1) [60]. Though almost all subtypes of calcium channels are highly expressed in the CNS [37], the “N-type calcium channels” play a key role in the release of the neurotransmitter from the afferent terminals within the spinal dorsal. Hence, they serve as a key therapeutic target during the treatment of analgesics [37]. Few other studies have also reported that the Cav2.2 expression in microglia is associated with the pathophysiology of neuropathic pain subsequent to spinal nerve injury [61]. As the “N-type calcium channels” are mainly expressed at nociceptive primary afferents synapse, an increased nociceptive function is observed subsequent to inflammation as well as neuropathy [62] and genetic deletion at this site reduces inflammatory pain behaviors [63]. Additionally, “N-type calcium channels” blockers diminish behavioral hyperalgesia and dorsal horn neurons’ hyperexcitability during inflammatory & neuropathic pain [64]. Few splice variants, especially e37a and e37b, of the “N-type calcium channel”, are reported to have different current densities and G-protein sensitivities and distinct electrophysiological properties. These variants are associated with the development of mechanical and thermal hyperalgesia at the time of inflammatory and neuropathic pain [65].
Migraine is also reported to involve neurogenic inflammation [66]. “P/Q-type calcium channels” is reported to play a key role in migraine. For instance, gain-of-function mutations in CACNA1A are a risk factor for familial hemiplegic migraine type 1 [36]. It enhances open channel probability and channel activation even at a lesser voltage [67]. Though the relation between the “P/Q-type calcium channels” and migraine is well established, their involvement in pain signaling is a topic of debate. The Nagoya mutant mouse with a loss-of-function mutation in “P/Q-type calcium channels” experiences less inflammatory pain phenotype [67]. Although complete “P/Q-type calcium channels” deletion causes hyposensitivity to neuropathic pain, they paradoxically enhance acute thermal nociception [68]. Thus, though “P/Q calcium channels” might be associated with pain signaling, their role is very limited in comparison to “N-type” as well asset-type calcium channels” [68].
All types of “L-type calcium channels” are present in the lymphocytes. Cav1.2 channels are highly expressed in the Type-2 helper T cells, where they play a vital role in modulating calcium signaling as well as cytokine production that is associated with allergic asthma [69]. Earlier studies have also reported that enhanced expression of Cav1.2 in murine microglial BV2 cells increases intracellular calcium concentration and TNFα and nitric oxide (NO) production, thereby supporting the association between Cav1.2 and CNS inflammation [70]. Few studies have reported that reduced Cav1.2 and Cav1.3 expression in dorsal root ganglion is associated with hyperexcitability during neuropathic pain [71]. One study reported that the expression of miR-103 gets reduced during neuropathic pain, which in turn enhances Cav1.2, thereby causing hyperexcitability as well as allodynia [72]. Cav1.2 post-translational modification in smooth muscle cells is thought to have a role in aberrant gastrointestinal smooth muscle motility after colonic inflammation and may contribute to intestinal dysmotility in inflammatory bowel disease patients (IBD) [73].
Few other studies have reported that-type calcium channels”, namely Cav3.2, is highly expressed in the primary sensory neurons and is responsible for modulating the excitation of neuron nearby peripheral axons, as well as the release of spontaneous neurotransmitter towards the central endings, thereby supporting their involvement in nociceptive processing, specifically in neuropathic pain [74]. The expression and function of Cav3.2 are reported to be modulated via various inflammatory mediators, for instance, bradykinin, hydrogen sulfide (H2S) as well as through the neuronal activity itself [47]. Cav3.2's enhanced activity is involved in colonic, bladder, and pancreatic pain, as well as regulating visceral inflammation. Inflammation and inflammatory pain are caused by VGCCs, and Cav3.2 T-type VGCC is a promising therapeutic target for the treatment of visceral inflammatory pain in patients with irritable bowel syndrome, interstitial cystitis/bladder pain syndrome, pancreatitis, and other conditions, as well as neuropathic pain [75].
5. DRUGS FOR REDUCING INFLAMMATORY PAIN CAUSED DUE TO CALCIUM CHANNELS REGULATION
Inhibition of calcium channels through various analgesics, for instance, pregabalin & gabapentin, are considered as the first-line treatment against neuropathic pain [76]. Protein kinase C (PKC) translocation caused by the pronociceptive peptides bradykinin and prokineticin 2, which are involved in both inflammatory and chronic pain, was dramatically reduced by gabapentin. After nerve injury, these drugs distinctly reverse the neural sensitization without distrusting the normal physiological pain [77]. Another drug, namely gabapentinoids, is reported to exercise their analgesic effect via blocking α2δ1 trafficking towards synaptic terminals within the spinal cord of neuropathic rats [78]. Nilvadipine, a calcium channel blocker, is reported to inhibit ocular inflammation in endotoxin-induced uveitis [79]. Amlodipine, lacidipine and verapamil also have an anti-inflammatory effect on calcium channels [80].
Agents that control the activity of high voltage-activated Calcium channels (HVCCs) relieve visceral pain in both experimental and clinical models. The antagonists of HVCCs and Transisent receptor potential cation channels receptor (TRPA1), Phα1β and CTK 01512-2 toxins showed an excellent response profile in the modulation of nociception and inflammatory process in the acute pancreatitis model in rats, without spontaneous movement of the animals [81]. Pimozide, a dopamine D2 receptor antagonist known as a conventional antipsychotic, and bepridil, a multi-channel blocker used to treat arrhythmia and angina, both show significant T-channel blocking activity [82]. In male rat pain models, inhibiting T-type channels lowers the excitability of peripheral nociceptive sensory neurons and reverses pain hypersensitivity. Spinal excitability and pain hypersensitivity are reduced by Z944, a T-type calcium channel antagonist [83]. Cav3.3 function inhibition or attenuation could be a useful therapy for trigeminal neuropathic pain. The features of calcium channel blockers as a vasodilator and inhibitors of muscular contractions, such as pernio, anal fissures, face wrinkles, and painful leiomyoma, are the main reasons for their usage in dermatology [84].
Irrespective of side effects, opioids serve as a key drug for the treatment of any pain [85]. After binding with opioids, the G protein-coupled µ-opioid receptor gets activated, which in turn lessens neuronal excitability as well as the release of a transmitter by inhibiting the function of “N-type calcium channels” and/or stimulating numerous potassium channels [86]. The “N-type calcium channels” blockers, namely, ziconotide and N-triazole oxindole, are also employed for the treatment of pain. Another drug, namely, TROX-1, is reported to reverse the allodynia induced via inflammatory-induced hyperalgesia nerve injury [87]. One study reported that collapsing response mediator protein-2 (CRMP-2) modulates the function of “N-type calcium channels”. CRMP-2 overexpression enhances calcium currents and, hence, “N-type calcium channels” expression. On the contrary, CRMP-2 knockdown shows contradictory effects [88].
Disruption of the interaction between “N-type calcium channels” and CRMP-2 decreases neurotransmitter release, calcium current, as well as inflammatory & neuropathic hypersensitivity [88]. Another “N-type calcium channels” blocker, namely NP078585 (a derivative of lomerizine and flunarizine), is reported to reduce inflammatory and neuropathic pain in a rat model [89]. As drugs produced from natural resources have less or no side effects, several studies have also proposed the usage of natural resources as drugs in various diseases, including pain [87, 88]. For instance, in 2013, Adams and the team proposed that ω-conotoxins, small disulfide-rich peptides isolated from the venom of the marine cone snail, function as “N-type calcium channels” inhibitors and block nociception during neuropathic and inflammatory pain models [90] (Table 1).
| Channel Types | Inhibitors | References |
|---|---|---|
| N-type calcium channels | Opioids | [82, 90] |
| Ziconitide and n-triazole oxindole | [92] | |
| CRMP-2 protein | [84] | |
| ω-conotoxins | [87] | |
| L-type calcium channels | Azelnidipine | [88, 91] |
| Nifedipine | [79, 91] | |
| Diltiazem | [86] | |
| T-type calcium channels | Ethosuximide, Mibefridl, KST5468, TTA-P2, TTA-A2 | [92] |
Another dihydropyridine derivative, namely azelnidipine, is annul-type calcium channels” blocker as well as antihypertensive. It causes minimum sympathetic nervous system stimulation and has a protective role in inflammation during atherosclerosis [90]. Nifedipine, an“L-type Calcium channels” blocker, reduces the expression of inflammatory cytokines, for instance, TNF-α and MIP-2 [88]. In another study, authors reported that Diltiazem, an “L-type calcium channels” blocker inhibits the formation of aneurysm through blood pressure-independent anti-inflammatory effects [89]. “T-type calcium channels “inhibition via ethosuximide, mibefridl, KST5468, TTA-P2 TTA-A2 have the antinociceptive ability within the inflammatory models, which in turn decreases paw edema as well as immune cell number. TRPA1 channel blocker, namely HC-030031, also work effectively during chronic inflammatory or neurogenic pain [90].
However, as nothing is perfect in this world, earlier several studies have reported that calcium channel could have an adverse effect on human health. For instance, diltiazem and verapamil may have a negative impact on cardiac conduction. Verapamil may cause constipation and nausea. A high dose of verapamil, diltiazem and nifedipine inhibits insulin secretion. Calcium channel blockers may also be responsible for causing hypertension and cancer. However, none of the calcium channel blockers have been reported to affect protein or lipid metabolism, severely [90-94]. Thus, calcium channel blockers may be consumed under the supervision of medical practitioners. Information obtained in the present study will be highly useful for understanding and treatment of pain caused by inflammation due to calcium ion channels.
CONCLUSION AND FUTURE PERSPECTIVE
In conclusion, the origin of every pain syndrome is inflammation and inflammatory responses. Various voltage-gated ion channels that are permeable to calcium ions mediate a number of signaling functions, including, neurotransmitter release, calcium-dependent enzyme activation and calcium-dependent gene expression modification that is associated with the pain behavior. Hence, understanding calcium signaling is an important step towards understanding the neural network activity associated with pain management. Results obtained revealed that these channels play a significant role during inflammatory and neuropathic pain. Thus, inhibition of these channels through various analgesics serves as a therapeutic treatment against inflammatory and neuropathic pain.
However, few of these inhibitors have various side effects, including cancer. Hence, these inhibitors may be consumed under the supervision of medical practitioners. Additionally, since the last few decades, technology advancement has been made to recognize drugs from phytochemicals against various diseases, including inflammation. As there are only a few calcium channel blockers for treating inflammatory and neuropathic pain, there is still scope for identifying them from various sources, including natural resources, during the drug discovery process. Authors believe that, as the computational high throughput screening process requires less time and capital, these computational approaches when combined with wet-lab approaches may hasten the process of drug discovery. Many avenues can be used to raise awareness of ongoing advancements. Reviewing clinical studies, looking into newly sponsored initiatives, and connecting with peers in the sector are just a few examples. Because pain continues to have such a negative influence on individuals, their environment, and the healthcare system, the present surge in pain research offers an exciting chance for really transformative advancements in pain management in the future.
LIST OF ABBREVIATIONS
| (CNS) | = Central Nervous System |
| (TRP) | = Transient Receptor Potential |
| (TRPV1) | = TRP vanilloid 1 |
| (TRPM8) | = Transient Receptor Potential Melastatin 8 channel |
| (VGKCs) | = Voltage Gated-potassium Channels |
| (DRG) | = dorsal root ganglion |
| IL-10 | = interleukin 10 |
| DRG | = Dorsal Root Ganglion |
| TG | = Trigeminal Ganglia |
| (IL-1) | = Interleukin 1 |
| TNF | = Tumor Necrosing Factor |
| AMPA | = Α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| NMDA | = N-Methyl-D-aspartic acid or N-Methyl-D-aspartate |
| LTCCs | = L-type Calcium Channels |
| NVGCC | = N-type Voltage-gated Calcium Channel |
| IBD | = Inflammatory Bowel Disease |
| PKC | = Protein Kinase C |
| HVCCs | = High Voltage Activated Calcium Channels |
| TRPA1 | = Transisent receptor potential cation channels receptor |
| CRMP-2 | = Collapsing response mediator protein-2 |
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
Dr. Senthilkumar Rajagopal is the Editorial Advisory Board Member for The Open Neurology Journal.
ACKNOWLEDGEMENTS
Declared none.

