All published articles of this journal are available on ScienceDirect.

An Atypical Case of POEMS Syndrome Associated with Autonomic Dysfunction
Authors Info & Affiliations
Abstract
A 52 year old patient showed, for two years, symptoms compatible with sensorimotor polyneuropathy preventing him from walking. An electroneuromyography found a demyelinating pattern suggesting Chronic Inflammatory Demyelinating Polyneuropathy (CIDP). The patient did not respond to corticosteroid therapy and later suffered sexual Dysfunction, Swelling, Lymphadenopathy, Hypotension, astrointestinal dysmotility, urinary retention and neuropathic pain. Analysis of the Cerebrospinal Fluid (CSF) revealed elevated protein levels and Computed Tomography (CT) scan found sclerotic bone lesions. High Vascular Endothelial Growth Factor (VEGF) levels and the results of Lambda light-Chain monoclonal gammopathy in urine protein Electrophoresis Suggested a Diagnosis of POEMS syndrome. The most striking feature, in this case, was the patient’s heightened and atypical polyneuropathy without axonal injury even after an extended period of time, and significant and atypical dysautonomia.
1. INTRODUCTION
POEMS syndrome is a rare paraneoplastic syndrome of hematological origin [1]. It is also known as osteosclerotic myeloma, Crow-Fukase syndrome and Takatsuki syndrome, and was first described in 1980 by Bardwick [1, 2]. POEMS is an acronym referring to several, but not all, pathological characteristics of the condition: polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal plasmacytic disorder and skin changes [1]. A few other non-represented characteristics are papilledema, extracellular volume overload, sclerotic bone lesions, thrombocytosis/erythrocytosis, increased levels of Vascular Endothelial Growth Factor (VEGF) and Castleman disease [2]. The physiopathology of POEMS syndrome is not well known but is commonly associated with chronic inflammation and overproduction of substances like VEGF, interleukin-1B, tumor necrosis factor-alpha and interleukin-6 [3].
POEMS syndrome’s clinical spectrum is fairly extensive. Peripheral nerves are a major target and the disorder is usually found in polyneuropathy patients with preserved autonomic function despite severe motor dysfunction [4-6]. Autonomic function preservation is typical of this syndrome and distinguishes it from other neuropathies such as diabetic neuropathy [3]. This paper reports the case of a POEMS syndrome patient who critically showed signs of dysautonomia, which strengthens the need for research on atypical cases of this disorder.
2. CASE REPORT
A 52 year old man of light brown skin from the state of Rio de Janeiro, Brazil, without any neurological comorbidities or family history, three years ago began showing bilateral gynecomastia and early-onset sexual dysfunction. Over the same period of time, the patient lost 20kg (44lbs), approximately 25% of his body weight. He also exhibited plantar paresthesia ascending to the upper limbs and culminating in a tetraparesis that prevented him from walking. After electroneuromyography, the patient was diagnosed with Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) and Corticotherapy was Initiated which Proved unsuccessful.
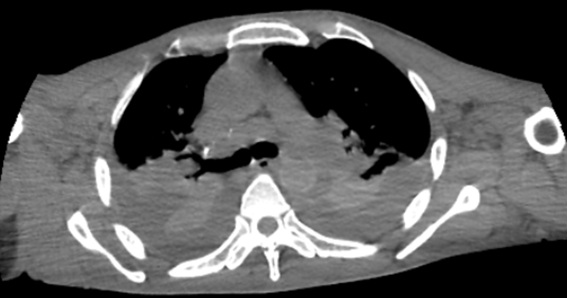
Subsequently and crucially, the patient sustained lower limb edema, ascites and bilateral pleural effusion (Fig. 1) and was admitted to the Gaffrée and Guinle University Hospital for diagnosis and treatment. During hospitalization, he suffered respiratory failure, requiring orotracheal intubation, dysautonomia with refractory arterial hypotension, xerostomia, urinary retention and constipation.
Physical examination revealed digital clubbing, axillary and inguinal lymph node enlargement with mobile and elastic lymph nodes, significant abdominal distension and no hepatosplenomegaly. Other findings included bilateral gynecomastia, symmetrical testicles of adequate size for his age and tetraparesis with a marked motor deficit in lower limb distal muscles. In addition to these symptoms the patient had global hyporeflexia, tactile and painful hypoesthesia, from the lower limbs to the distal region of the thigh and apalesthesia. A fundoscopic examination found no sign of papilledema.
Initial laboratory tests revealed thrombocytosis. No changes were observed in the thyroid hormones, glycated hemoglobin, cyanocobalamin, HIV, syphilis, and hepatitis serologies. Sexual dysfunction was reported. The prolactin level mildly increased at 34 ng/ml (normal <15); luteinizing hormone and follicle-stimulating hormone levels were normal. Testosterone was not tested.
Analysis of the patient’s Cerebrospinal Fluid (CSF) found increased protein and glucose levels at 134 mg / dL (normal <45) and 83mg/dL (normal 40-70), respectively. Cytometry found 2 leukocytes that were 100% mononuclear.
Electroneuromyography revealed sensorimotor peripheral polyneuropathy with a demyelinating pattern without axonal loss. There was no conduction block. Conduction velocities ranged from 7.5 to 13.5 m/s and response amplitudes ranged from 0.1 to 0.2 mV. Phrenic nerve analysis also revealed a demyelinating pattern.
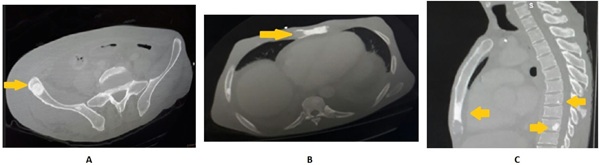
A transthoracic echocardiogram confirmed mild pericardial effusion, moderate left-ventricle systolic dysfunction and diastolic dysfunction. A Computed Tomography (CT) scan revealed lymphadenopathy in the lower paratracheal, subcarinal and para-aortic chains in addition to sclerotic bone lesions in the lower third of the sternum body, left anterior acetabulum, right ilium and T9, T11 and T12 vertebral bodies (Fig. 2).
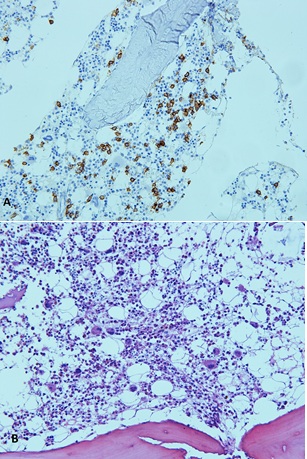
The results of the axillary lymph node biopsy were consistent with a reactional pattern (Fig. 3). Bone marrow biopsy revealed 50% cellularity with the three hematopoietic series, along with 10% plasma cells (Fig. 4). No peripheral nerve biopsy was performed.
Peripheral blood protein electrophoresis showed homogeneous distribution of immunoglobulins and urine protein electrophoresis demonstrated monoclonal peak at lambda light chain.
VEGF concentration was 300 times the reference value.
The association of demyelinating polyneuropathy with lambda light-chain monoclonal gammopathy in urine electrophoresis, along with sclerotic bone lesions, increased VEGF, extracellular volume overload, lymph node enlargement, endocrinopathy, thrombocytosis, weight loss and digital clubbing confirmed the diagnosis of POEMS syndrome.
After diagnosis, the patient underwent chemotherapy with cyclophosphamide, dexamethasone and interferon. This regimen succeeded in partially improving motor function but the patient continued to suffer from neuropathic pain and significant dysautonomia and the ensuing tachycardia, xerostomia, urinary retention, heat intolerance, bowel constipation and refractory hypotension. Noradrenaline was then used to maintain effective hemodynamic stability. The patient passed away due to pneumonia, three years after the onset of the first symptoms and 45 days after the initial treatment.

3. DISCUSSION
POEMS syndrome diagnosis is based on major and minor criteria. The mandatory major criteria are polyneuropathy-typically demyelinating, sensorimotor, progressive and distal-and monoclonal gammopathy. There are three main secondary criteria, any one of which suffices for diagnosis purposes: Castleman disease, sclerotic bone lesions or elevated VEGF. There are six minor criteria, any one of which suffices for diagnosis purposes: organomegaly, extravascular volume overload, endocrinopathy, skin changes, papilledema or thrombocytosis/polycythemia. Clubbing, weight loss, pulmonary hypertension, diarrhea and low vitamin B12 levels can also be included, however, none of these are considered the diagnostic criteria [1, 4].
The patient showed four out of five major criteria and four minor ones. Initially diagnosed with CIDP, he did not respond to corticotherapy. A detailed history and physical examination indicated that the CIDP diagnosis was insufficient as other organs and systems had been affected.
POEMS syndrome neuropathy usually begins with lower limb distal pain and paresthesia followed by motor disorders. The latter can be predominantly distal or, in the case of CIDP, show symmetrical proximal distribution [3, 7]. Both conditions involve albuminocytologic dissociation in CSF and electrophysiological evidence of demyelinating f wave prolongation, slow conduction velocities and temporal dispersion [5]. The electroneuromyography of POEMS syndrome patients exhibits heightened axonal loss [2]. They maintain autonomic dysfunction despite significant motor dysfunction [6].
Although the reported patient’s clinical conditions were consistent with the syndrome, his neuropathy pattern was unusual. Electroneuromyography revealed a demyelinating sensorimotor polyneuropathy pattern. The patient also showed dysautonomia signs that began with sexual dysfunction and progressed to heat intolerance, severe arterial hypotension, tachycardia, urinary retention, chronic constipation and xerostomia [8]. The autonomic dysfunction pattern is unusual for POEMS syndrome and suggests widening diagnostic possibilities in the field of demyelinating polyneuropathy.
CONCLUSION
Polyneuropathy is one of the two major mandatory criteria for POEMS syndrome diagnosis. In these patients, neurological damage usually follows a demyelinating pattern, with greater axonal lesions and autonomic preservation. However occasionally, atypical cases without axonal lesions and significant autonomic dysfunction are observed. This report is significant as this atypical presentation draws attention to the syndrome’s diagnosis. This report will hopefully stimulate research on the rare POEMS syndrome patients whose polyneuropathy is of unknown etiology and leads to less underreporting.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
STANDARD OF REPORTING
CARE Guideline and methodology were followed.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We would like to thank the Seventh Infirmary of Gafrée and Guinle University Hospital for being the cradle of so much learning, love of medicine and welcoming patients.

