All published articles of this journal are available on ScienceDirect.

Inflammasome-Mediated Inflammation in Neurodegenerative Diseases
Abstract
Inflammasomes are protein platforms consisting of multiple proteins. The biological function includes the activation of caspase-1, leading to the maturation of IL-1β and IL-18. These pro-inflammatory cytokines promote fundamental inflammatory processes in numerous infectious diseases. The inflammasome-mediated inflammation has become increasingly important in central nervous system disorders. In neurodegenerative disorders, significant contributors to disease progression include neuroinflammation and inflammatory cascades initiated by the inflammasome protein complex. This review discusses the recent progress of research on inflammasome associated with neurodegenerative disorders.
1. INTRODUCTION
Neurodegenerative disorders are characterized by the progressive structural and functional degeneration of nerve cells in the Central Nervous System (CNS) [1]. Most neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Multiple Sclerosis (MS), Amyotrophic Lateral Sclerosis (ALS), and epilepsy, are associated with chronic neuroinflammation [2, 3] and increased levels of cytokines and activated immune cells [4].
Inflammasomes expressed in phagocytes, including macrophages and dendritic cells, activate caspase-1 in response to pathogenic infections and tissue damages [5]. The activation of inflammasome has been linked to different diseases including viral infections [6], diabetes [7], hypertension [8], and rheumatoid arthritis [9]. In CNS, inflammasomes play a pathogenic role in infectious conditions such as pneumococcal meningitis [10], Toxoplasma gondii infection [11], murine Japanese encephalitis [12], and HIV/AIDS [13]. Recently, inflammasomes have also been linked to neurodegenerative diseases. In this review, we discuss the recent research progress on the activation and function of inflammasomes in neurodegenerative disorders.
2. INFLAMMASOMES
Inflammasomes are multimeric protein complexes comprising of Pattern Recognition Receptor (PRR), an apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) adaptor protein, and procaspase-1 [14]. The recognition of pathogen-associated molecular patterns and danger-associated molecular patterns by PRR triggers the assembly of the inflammasome complex [15]. Inflammasome oligomerization leads to self-cleavage of procaspase-1 to generate activated caspase-1, which is essential for the maturation of interleukin-1β (IL-1β) [16] and interleukin-18 (IL-18) [17]. PPRs are divided into four distinct classes: Toll-Like Receptors (TLRs), C-type Lectin Receptors (CLRs), Retinoic acid-inducible gene I-Like Receptors (RLRs), and Nucleotide-binding oligomerization domain-Like Receptors (NLRs). It has been reported that few NLR family members including NLRP1, NLRP3, NLRC4, Pyrin, and AIM2 are able to assemble the inflammasome complex in vivo [18-20]. Of those, NLRP3 inflammasome has been extensively studied and is linked to neurodegenerative disorders. Inflammasomes are mainly involved in the innate immune response, contributing to neuroinflammatory damage (Fig. 1). The inflammasome mediates secretion of IL-1β and IL-18 which induces the fundamental inflammatory events in neuroinflammation [21].
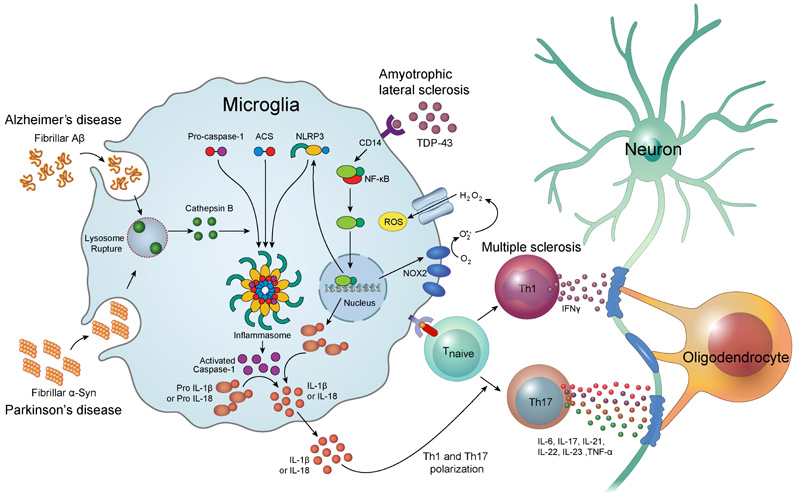
3. ALZHEIMER’S DISEASE
Alzheimer’s Disease (AD) is a progressive neurodegenerative disease characterized by dementia and memory loss [22]. The pathological hallmarks of AD are the extracellular deposits of amyloid plaques and intracellular accumulation of Neurofibrillary Tangles (NFTs) [23]. Although there are numerous possible etiologies for AD, the exact mechanisms for the onset remain unclear. Recently, neuroinflammation has emerged as an important risk factor in the development of AD. In CNS, microglia play a pivotal role in the inflammatory reaction [24]. In an AD patient's brain, the microglia are seen gathered around the amyloid β (Aβ) plaques [25] and induce massive neuronal cell death through secretion of tissue necrosis factor α (TNF-α) [26].
Helle et al. first reported that NLRP3 inflammasomes can be activated by fibrillar Aβ. Phagocytosis of fibrillar Aβ by activated microglia induces lysosomal damage, resulting in the leakage of cathepsin B. This study revealed that cytoplasmic cathepsin B activates NLRP3 inflammasome and induces the release of IL-1β by microglia [27]. The study by Heneka et al. reported that knockdown of NLRP3 decreases the accumulation of Aβ, and prevents the behavioral and cognitive dysfunction in the aged APP/Presenilin-1 (PS1) transgenic mice model of AD. APP/PS1/NLRP3-/- mice also showed decreased accumulation of Aβ plaque in the hippocampus [28]. Other evidence has suggested that activation of the nuclear factor-kappa B (NF-κB) pathway has a critical role in the activation of NLRP3 inflammasome [29]. Shi et al. showed that artemisinin, a known antimalarial drug, significantly inhibited the activation of NF-κB and NALP3 inflammasome, and reduced the amyloid plaque deposition in the cortex and hippocampus in APPswe/PS1dE9 transgenic mice [30].
It has also been reported that NLRP1 contributes to the age-related neuronal loss. The expression of NLRP1 was observed to increase after 6 months in APP/PS1 mice; however, knockdown of NLRP1 decreases the neuronal cell death and rescues early cognitive deficits [31]. Kaushal et al. showed that NLRP1 inflammasome activates caspase-1, which subsequently activated caspase-6, in human primary neurons treated with serum deprivation and benzylated ATP [32]. Caspase-6 is known as a key effector of apoptosis. Numerous other evidences reveal that caspase-6 activity is strongly associated with AD pathologies. LeBlanc et al. reported that caspase-6 is responsible for the increased levels of Aβ in primary cultures of human neurons [33]. It was also reported that the activated caspase-6 is exceedingly observed in the brain of both sporadic and familial AD [34, 35].
Tau protein can be cleaved by caspase-3 at Asp421 [36] and this truncated tau (TauΔCasp3) induces cell death in the primary hippocampal neurons [37]. Caspase-6 also cleaves the tau at Asp402 [38] and Asp13 [39] in vitro. Another study showed that the activated caspase-6 and TauΔCasp6 were highly observed in neuropil threads, NFTs, and neuritic plaques of end-stage AD brain [38].
4. PARKINSON’S DISEASE
Parkinson’s Disease (PD) is the second most common neurodegenerative disorder, characterized by progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) located in the midbrain [40]. The pathological hallmark of PD is Lewy bodies composed of misfolded α-synuclein (α-Syn) aggregates [41]. Pathogenesis of PD is still unclear, but accumulating evidence indicates that neuroinflammation may act as a risk factor for the development of PD. Significantly increased levels of pro-inflammatory cytokines, including IL-1, IL-2, IL-6, and TNF-α, were detected in the serum and Cerebrospinal Fluid (CSF) of PD patients [42]. It was reported that the use of non-steroidal anti-inflammatory drugs reduces the risk of PD onset [43, 44].
Several studies have shown that α-Syn activates microglia in vitro and in vivo [45-47]. The exposure of monocytes to fibrillar α-Syn induced the secretion of IL-1β via caspase-1 activation and the up-regulation of NLRP3 [48]. This study also showed that phagocytosis of fibrillar α-Syn by monocytes leads to the release of cathepsin B and production of Reactive Oxygen Species (ROS), which are known activators of NLRP3 inflammasomes. A recent study reported that α-Syn can be cleaved directly by caspase-1 in vitro [49]. The level of cytotoxicity in neuronal cells correlated with the level of truncated α-Syn, but inhibition of caspase-1 activity rescued the α-Syn-induced cytotoxicity.
MicroRNA-7 (miR-7) is a direct regulator of α-Syn in post-translational modification. Li et al. reported that miR-7 has a neuroprotective effect in the 1-methyl-4-phenylpyridinium (MPP+) mediated PD model [50]. In this study, the MPP+-elicited apoptotic effects on neurons were significantly reduced by miR-7. Another study showed that NLRP3 inflammasomes are activated in the midbrain of α-Syn-overexpressed A53T transgenic mice, but subsequent transfection of miR-7 significantly inhibited the A53T α-Syn-induced the upregulation of NLRP3 [51].
ATP13A2 gene, also called Park9, encodes a transmembrane lysosomal p5-type ATPase (ATP13A2) which is involved in the stabilization of lysosome membrane structure. It has been reported that ATP13A2 is highly expressed in the SNc [52]. Other studies reported that deficiency of ATP13A2 leads to the accumulation of α-Syn in neuronal cells [53], and the mutation of ATP13A2 gene causes an early-onset PD [54]. The study by Qiao et al. reported that knockdown of ATP13A2 in primary astrocytes causes increased secretion of pro-inflammatory cytokines (TNF-α and IL-6), and decreased the production of anti-inflammatory cytokines (IL-4 and IL-10). In addition, the downregulation of ATP13A2 increased the expression of astrocytic cathepsin B, which subsequently induces the activation of NLRP3 inflammasome [55].
5. MULTIPLE SCLEROSIS
Multiple Sclerosis (MS) is an autoimmune disease characterized by the progressive loss of myelin sheaths of neurons. T lymphocytes play a pivotal role in the pathogenesis of MS [56]. It has been reported that high numbers of T cells, especially myelin-specific autoreactive T cells, are present in the peripheral blood of MS patient [57]. Immune cell infiltration into the CNS is tightly controlled by the Blood-Brain Barrier (BBB). Nevertheless, many immune cells are able to cross the BBB in neuroinflammatory diseases. The etiology of MS is still not known, but it is thought that the activated myelin-specific T cells cross the BBB and trigger the recruitment of other inflammatory cells, which consequently lead to the destruction of the myelin sheath [58].
T helper type 1 (Th1) cells are the main effector cells, which activate the macrophages via interferon-gamma (IFN-γ). Activated macrophages promote neuroinflammatory events by releasing inflammatory mediators including cytokines, ROS, nitric oxide and glutamate, which then induce tissue damage [59]. IL-17-producing effector T helper cells, called Th17 cells, have emerged as key mediators of MS. Th17 cells produce the effector cytokines including IL-6, IL-17, IL-21, IL-22, IL-23 and TNF-α, which induce the inflammatory reactions [60].
Previous studies have shown that NLRP3 inflammasome contributes to the development of MS. It was observed that the expression of caspase-1 is significantly increased in the Peripheral Blood Mononuclear Cells (PBMC) of MS patients [61]. Another study reported that the mRNA expression of inflammasome associated molecules, including NLRP3, caspase-1, IL-1β, is increased in the PBMC of MS patients as compared to the healthy control group [62]. In active demyelinating lesions of MS, reactive astrocytes and infiltrated perivascular macrophages express IL-1β and NLRP3 inflammasome components including NLRP3, ASC, and CASP1 [63].
Cuprizone is known to cause extensive demyelination in the corpus callosum [64]. Jha et al. reported that the expression of Nlrp3 is increased in the cuprizone-induced demyelination model [65]. This study also showed that demyelination and microglial infiltration are significantly reduced in the cuprizone-treated Nlrp3-/- mice, Casp1-/- mice and IL-18−/− mice; however, no alteration was observed in IL-1β−/− mice. Experimental Autoimmune Encephalomyelitis (EAE) is a widely-used rodent model for MS [66]. It is known that macrophage and microglia were involved in the development and progression of EAE [67-69]. However, the study by Vainchtein et al. reported that the role of macrophages and microglia is different in acute EAE [70]. This study showed that infiltrated macrophages were highly immune reactive, while microglia were only weakly immune activated during acute EAE. In the EAE model, Nlrp3-/- mice displayed reduced severity of EAE, and significant reduction of the inflammatory cells infiltration including macrophages, dendritic cells, CD4+, and CD8+ T cells [71]. IFN-γ and IL-17 are key pro-inflammatory mediators in the development of EAE, and their levels were significantly reduced in the Nlrp3-/- mice and IL-18−/− mice. Another study showed that Asc−/− mice and Nlrp3−/− mice were resistant to the development of EAE. In addition, both EAE-induced Asc−/− mice and Nlrp3−/− mice showed significantly reduced numbers of CD4+ T cells in the spinal cord and brain [72]. These findings suggest that NLRP3 inflammasome is required for the development of EAE.
6. AMYOTROPHIC LATERAL SCLEROSIS
Amyotrophic Lateral Sclerosis (ALS) is a fetal neurodegenerative disorder characterized by progressive degeneration of the upper and lower motor neurons [73]. Astrocytes and microglia play major roles in the disease progression of ALS. The aggregation of mutant Superoxide Dismutase 1 (SOD1) is the pathologic hallmark of familial ALS (fALS) [74]. It has been reported that astrocytes expressing mutated SOD1 result in the death of primary motor neurons through the activation of a Bax-dependent pathway [75]. Another study showed that microglia in SOD1G93A transgenic mice carrying the human SOD1 mutant gene induced the motor neuron death via the NF-κB dependent mechanism [76].
There is accumulating evidence suggesting that inflammasome is involved in the progression of ALS. The expression of NLRP3 inflammasome components and IL-1β were increased in the SOD1G93A mouse model, as well as in spinal cord tissue of human sclerosis ALS (sALS) patients [77]. Another study showed that the IL-18 expression was increased in the cerebral tissue of sALS patients as compared to an age-matched control group [78]. Symptoms of cognitive and behavioral impairment in the later stages of ALS are believed to be the results of neurodegeneration in the subcortical areas. Massive dendritic swelling and neuronal loss were detected in SOD1G93A mice. In addition, the accumulation of misfolded SOD1 protein and autophagy markers was observed in the anterodorsal nucleus of the anterior thalamus [79]. This study also showed that the expression of NLRP3 and ASC was significantly up-regulated in the anterodorsal thalamic nucleus of SOD1G93A mice.
TAR DNA binding protein (TDP-43) is the insoluble multifunctional nucleic acid binding protein which has an important role in neuronal RNA metabolism related to neuronal development and synaptic function [80]. TDP-43 is normally located in the cell nucleus; however, enhanced deposition of TDP-43 in the cytoplasm is observed in ALS [81]. A recent study reported that the interaction of TDP-43 with CD14 receptor in microglia triggers the activation of the microglial NF-κB, AP-1 and NLRP3 inflammasome pathways, leading to the production of TNF-α and IL-1β [82]. This study showed that TDP-43 induced the up-regulation of NLRP3 mRNA and activation of caspase-1, but did not alter the mRNA expression of NLRP1, NLRP2, AIM2, and NLRC4. TDP-43 also induced the up-regulation of NADPH oxidase 2 (NOX2), which is known an important source of ROS [83, 84]. ROS have been suggested as the key triggers of NLRP3 inflammasome activation [85].
7. EPILEPSY
Epilepsy is a chronic neurological disorder that is characterized by spontaneous recurrent seizures accompanied by cognitive impairment and psychiatric disturbances [86]. Accumulating evidence suggests that the unbalanced regulation of neuroinflammation plays a key role in the development of seizures and epilepsy [87]. Increased levels of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, are detected in the brain of an epilepsy model [88], and in the serum and CSF of epilepsy patients [89, 90]. In addition, the microglia activation is correlated with the increased expression of pro-inflammatory cytokines in the epileptic brain [91]. Microglia display both neurotoxic and neuroprotective effects in CNS disease [92]. Recent studies have shown that myeloid infiltrates, including monocyte and macrophages, and astrocytes exacerbate the neuroinflammatory status, whereas microglia play a protective role during early epileptogenesis [93, 94].
Several studies have reported that NLRP3 inflammasome can be up-regulated in an epilepsy model. The expression of cleaved IL-1β and hippocampal NLRP3 inflammasome components was elevated in a rat brain after Status Epilepticus (SE). On the other hand, siRNA knockdown of NLRP3 reduced the levels of IL-1β, IL-18, and caspase-1 expression, and inhibited hippocampal neuronal loss [95]. Similarly, the levels of IL-1β, NLRP3, and caspase-1 expression were up-regulated in a Kainic Acid (KA)-induced epilepsy model; however, curcumin suppressed the protein expression of IL-1β, NLRP3 and caspase-1, and reduced neuronal loss in the hippocampus [96]. NLRP3 inflammasome can be activated by oxidative stress [97]. The concentration of oxidative stress markers, including nitrite and malondialdehyde (MDA), and the expression of IL-1β, NLRP3, and caspase-1 were increased significantly in a KA-induced Temporal Lobe Epilepsy (TLE) model. In contrast, the high antioxidant activity, including glutathione (GSH), superoxide dismutase (SOD), and catalase, was decreased significantly [98]. Huperzine A (Hup-A), a natural acetylcholinesterase inhibitor, has been used for the treatment of AD because of its neuroprotective effects [99]. In the KA-induced TLE model, treatment with Hup-A reduced the nitrite and MDA concentrations, as well as the expression of IL-1β and caspase-1, while it increased the SOD and catalase activities.
Recent evidence also suggests that NLRP1 contributes to the pathogenesis of TLE. The increased expression of NLRP1 and caspase-1 was observed in the hippocampus of mesial TLE patients. In an amygdala kindling-induced TLE rat model, siRNA knockdown of NLRP1 reduced neuronal loss and caspase-1 expression and attenuated the seizure frequency and severity [100]. Similarly, the expression of inflammasome components, including NLRP1, ASC, and csapase-1, and inflammatory cytokines, including IL-1β, IL-18, IL-6, and TNF-α, increased in the brains of a pentylenetetrazole-induced epilepsy model. On the other hand, treatment with sinomenine, an anti-rheumatic alkaloid, suppressed the expression of NLRP1 inflammasome components and inflammatory cytokines [101].
CONCLUSION
In this review, we focused on the role of inflammasomes in neurodegenerative disorder. Each neurodegenerative disease has its own prognostic characteristic and symptomatic patterns. However, the up-regulated inflammatory response is common to all diseases. Inflammasomes especially play an important role in the initiation and progress of neuroinflammation. In AD, the amyloid plaques and neurofibrillary tangles activate inflammasomes in the microglia, astrocytes and neuron itself. As a consequence, the level of IL-1β increases in the CNS of AD patients, thereby promoting neuroinflammation. In Parkinson’s disease, α-synuclein aggregation activates the inflammasome complex in microglia. Enhanced NLRP3 inflammasome activation and up-regulated caspase-1 were detected in the postmortem MS brain. Increased expression of NLRP3 inflammasome components and IL-1β was observed in ALS animal models, as well as human CNS tissue. The up-regulated expression of NLRP1 and NLRP3 inflammasomes was detected in the brain tissue of an epilepsy model. As discussed in this paper, inflammasome is a major contributor to neuroinflammation in neurodegenerative disorders. However, only a few inflammasomes have been characterized. Better understanding the role of the inflammasome in neuroinflammation will provide more information to investigate the pathogenesis of neurodegenerative disorders.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2015R1D1A3A01019515).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors would like to thank Dong-Su Jang, MFA, (Medical Illustrator, Medical Research Support Section, Yonsei University College of Medicine, Seoul, Korea) for his help with the illustrations.

