All published articles of this journal are available on ScienceDirect.

Cutaneous and Mixed Nerve Silent Period Recordings in Symptomatic Paroxysmal Kinesigenic Dyskinesia
Authors Info & Affiliations
Abstract
Objective:
The underlying neurophysiologic mechanism responsible for secondary paroxysmal kinesigenic dyskinesia (PKD) is still unclear. Here, we study the pathogenesis of PKD in two patients with a demyelinating lesion in the spinal cord.
Methods:
Electromyogram recordings from affected arms of two patients with spinal cord lesions presenting PKD were compared with our laboratory standards. The cutaneous silent period (CuSP), mixed nerve silent period (MnSP) and coincidence period (CiP), defined as the common period between the CuSP and MnSP, were recorded.
Results:
A large decrease in the MnSP and disappearance of the CiP were observed in our patients, which was secondary to simultaneous extinction of the third portion of the MnSP, while the CuSP was normal. The MnSP and CiP were normal after recovery.
Conclusions:
Our results demonstrate that the third portion of the MnSP and the CuSP do not correspond to the same physiologic process. These findings suggest that PKD patients have abnormal spinal interneuron integration.
INTRODUCTION
Dystonia is a movement disorder characterised by sustained muscle contractions, frequently causing twisting and repetitive movements or abnormal postures. Classification is based on the affected body region, the age of onset or etiology [1]. Paroxysmal kinesigenic dyskinesia (PKD) is characterised by short attacks of dystonia or chorea which are precipitated by sudden voluntary movement. The attacks, which may occur up to 100 times per day, can also be precipitated by noises or hyperventilation. Attacks are usually unilateral and short, lasting seconds or minutes. Symptoms may be preceded by an increased tactile sensitivity in the limbs and may be limited to one side of the body or to a single limb. PKD can be divided into idiopathic and secondary (or symptomatic) cases [2]. Symptomatic PKD is observed in many different diseases including stroke, multiple sclerosis, cryptogenic myelitis and arterio-venous malformation, and the reported lesions are widely distributed from the cerebral cortex to the spinal cord [3-6].
The underlying neurophysiologic mechanism responsible for PKD is still unclear. While most lesions responsible for dystonia involve the caudate nucleus, the putamen, the globus pallidus and the thalamus [7, 8], it has been proposed that PKD is likely to be caused by a transversely spreading ephaptic activation of damaged axons at any level in a motor path [9].Nevertheless, more recent studies based on imagery suggested basal ganglia involvement in PKD pathogenesis [2].
A silent period in an electromyography (EMG) is a temporary, relative or absolute decrease in electromyographic activity of a contracted voluntary muscle occurring in response to a stimulus [10, 11]. A silent period can be elicited by a mechanical or electrical peripheral stimulus, electrical cortical stimulus, and transcranial magnetic stimulation of the brain. A peripheral silent period can be reproducibly triggered by cutaneous stimulation (CuSP) or mixed nerve stimulation (MnSP). Most investigators agree that the afferent impulses that generates the CuSP could be due to high-threshold thinly myelinated fibers [10-15]. The MnSP seems to be made up of three different parts including collision of antidromic with orthodromic motor impulses, Renshaw cell inhibition activated by antidromic motor volley and activation of high-threshold cutaneous fibres within the mixed nerve [16, 17]. The latter part has been suggested to share a common inhibitory pool with the CuSP [18]. While the underlying circuitry of the MnSP and CuSP remain uncertain, latencies indicate a spinal oligosynaptic inhibitory reflex [15, 19]. The aim of the present study was to explore physiologic spinal interneuron mechanisms in patients with PKD by analysing the CuSP and MnSP.
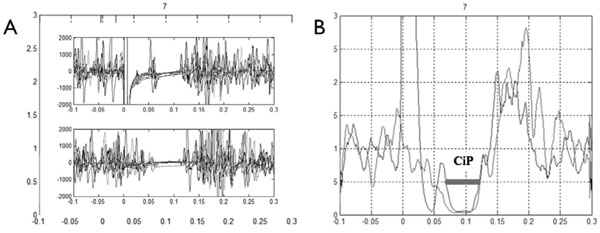
Electromyogram of a healthy subject. (A) Superposition of the eight replicate traces (Top panel, mixed nerve silent period; bottom panel, cutaneous silent period on the bottom). (B) Coincidence period (CiP) visualized on rectified normalized averaged trace.
PATIENTS AND METHODS
Patient 1:
A 55-year-old woman was admitted in March 2006 for tetraplegia with bilateral pyramidal signs and a deficit of sense of vibration that had developed three weeks earlier. Cerebrospinal fluid showed a normal cellularity and protein content, gamma globulin fraction was normal and oligoclonal bands were absent. Blood analyses were normal. Cerebral magnetic resonance (MR) was normal. Spinal cord MR showed hyperintensities on the T2 sequence from level C2 to C6 with no enhancement after injection of gadolinium. Other complementary investigations excluded multiple sclerosis, neuromyelitis optica, sarcoidosis or vasculitis. Idiopathic acute transverse myelitis was diagnosed. Two days later the patient complained of involuntary tonic contractions. They occurred up to 60 times per day, involved the right side, mainly the muscle flexors of the fingers and the forearm, and were preceded by parasthesias of the hand. PKD was precipitated by voluntary movement and hyperventilation, and was painful. Each episode lasted 30 seconds to one minute. Treatment with gabapentine (600mg three time a day) was effective after two weeks. Myelitis was successfully treated with intravenous methylprednisolone and intravenous pulses of cyclophosphamide.
Patient 2:
The second patient was a 58-year-old woman who had suffered from secondary progressive multiple sclerosis for 15 years with an expanded disability status scale (EDSS) score of 6.0. She complained of PKD in her right lower limb. These attacks lasted approximately one minute, occurred as often as 30 times a day and were precipitated by hyperventilation. Cerebral MR was unchanged but spinal cord MR showed a new recent T2-weighted hyper intensity at the C6/C7 level without enhancement after injection of gadolinium. Relapse was diagnosed and treatment with gabapentine (800 mg twice daily) was also effective in 10 days.
The study was conducted in full accordance with the World Medical Association’s Declaration of Helsinki.
Data Collection:
Neurophysiological investigations were performed with standard electrodiagnostic equipment (Viking IV, Nicolet®) set at 500-1000 µV/div sensitivity and with 2 to 2000 Hz bandpass with sampling at 5000 Hz. The CuSP was evoked by 0.2 msec constant current square wave electrical stimuli which were applied with a ring electrode to median sensitive fibers on the index finger. Stimulus intensities were set at 15 times the subjective sensory threshold determined at the start of the testing session for each subject. The MnSP was evoked from the median nerve on the wrist using a bar electrode (30 mm inter-electrode distance) with 0.3 msec stimulus duration, and intensities were 2 times the motor threshold. EMG activity was recorded from the abductor pollicis brevis muscle belly and its tendon using surface electrodes (10 mm diameter). Subjects were asked to maintain isometric contraction; intensity of the contraction was adjusted to about 50% of maximum strength based on audio feedback of muscle activity. Height traces were recorded for each subject. EMG was recorded in 400 ms trials divided into two parts: 100 ms before stimulation (as baseline EMG activity) and 300 ms after the stimulus, and the EMG activity, which included the silent period, was recorded. Neurophysiological investigations were performed for both patients during the PKD period (but they had no dystonic spams during the time of recording) and post-recovery for Patient 1. Patient 2 was not available for further examination during post-recovery.
Data Analysis:
Offline analysis was done using Matlab® software. A Butterworth 50-1000 Hz filter and weighted moving average (8ms window) were applied. Next, EMG traces were full-wave rectified and normalised for each condition recorded. At least 8 traces were averaged and a single rectified normalised averaged trace was obtained for each patient. The CuSP and MnSP were automatically defined as the time periods when the EMG signal dropped below 50% of the baseline preceding the stimulus. Three parameters were recorded. The first and second, the CuSP and MnSP, were classically defined as the time between stimulation and the end of the silent period. The third was a new index named coincidence period (CiP), defined as the common period between the CuSP and MnSP as shown in Fig. (1B).
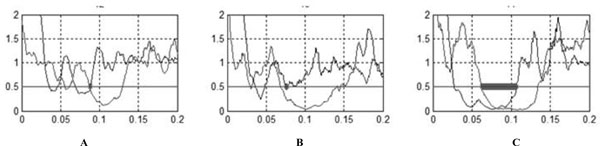
Rectified normalized averaged traces of patients.
(A) Patient 2.
(B) Patient 1 during paroxysmal kinegenic dystonia period.
(C) Patient 1 after recovery.
RESULTS
The CuSP, MnSP and CiP were systematically and easily obtained for healthy subjects. Waveform characteristic and reproducibility of the recordings taken for a healthy subject are illustrated in Fig. (1A). The constancy of inhibition during the silent period and the abrupt return of muscle activity are evident. Table 1 shows laboratory standards and values obtained with patients (with Patient 1 during PKD and after recovery). Several abnormalities were observed for both patients during PKD (Fig. 2): first a large decrease in the duration of the MnSP, second a small increase in the duration of the CuSP and third, disappearance of the CiP. Disappearance of the CiP was secondary to simultaneous extinction of the third portion of the MnSP while the CuSP was normal. Furthermore, a normal MnSP (106.8 msec) and CiP (44.8 msec) were observed in Patient 1 after recovery.
DISCUSSION
The protocols, parameters of stimulation and data collection methods used here were similar to those reported in the literature. Our laboratory standards were comparable with those previously described [17, 18].
Prolongation of the CuSP has already been described in patients with brachial dystonia compared to healthy subjects and could be attributed to abnormal timing in supraspinal inhibitory modulation of muscle activity [20]. Although factors determining the CuSP duration are poorly understood, spinal segmental interneurons are likely involved [21], and there is experimental evidence that Renshaw cells may also play a major role in the generation and maintenance of the CuSP [15].
An abnormal extinction of the CiP during PKD was observed in both patients. This may be explained by a dissociation between the presence of the CuSP and the disappearance of the last portion of the MnSP. This result is in apparent contradiction with current understanding, according to which the third portions of the MnSP and CuSP corresponds to the same physiologic process, namely activation of thinly myelinated fibers causing spinal interneuron oligosynaptic inhibition of motor neurons.
On the one hand, conservation of the CuSP shows that a single stimulation of thinly myelinated fibers is still able to inhibit the lower motor neurons. On the other hand, extinction of the third portion of the MnSP shows that simultaneous activation of orthodromic thinly myelinated fibers and antidromic motor impulses cannot generate late oligosynaptic inhibition of the lower motor neurons.
Duration (msec) of MnSP, CuSP and CiP for patients 1 and 2.
| MnSP | CuSP | CiP | |
|---|---|---|---|
| Laboratory Standards | 121.2 ± 18.75 | 123.2 ± 6.5 | 44.2 ± 13.5 |
| Patient 1 | 76.6 | 140.8 | 1.2 |
| Patients 2 | 88.4 | 129 | 1 |
| Patient 1 after recovery | 106.8 | 134.2 | 44.8 |
This dissociation may suggest different interpretations:
First, the population of cutaneous fibers activated by 2 times the motor threshold and 15 times the sensory threshold may be different. However, this seems unlikely because all calculations suggest that these afferent impulses of the CuSP and third portion of the MnSP are carried by slowly conducting fibers at a rate of 10 to 15 m/s, which is the rate of conduction of delta fibers [17, 22]. Furthermore the CuSP and the later portion of the MnSP were absent or shortened on the affected side of 4 patients with cervical syringomyelia and in 2 patients with peripheral small fiber neuropathy [16, 23].
The dissociation could also be explained by the activation of a second population of interneurons by the antidromic motor impulse, which would inhibit orthodromic activation of thinly myelinated fibers. These results tend to prove that patients with PKD have abnormal spinal interneuron integration.
This dysfunction of spinal inhibition seems to be related to the spinal lesion. This hypothesis is supported by the observation that after the clinical, radiological and neurophysiological recovery of Patient 1, the EMG showed a normal pattern of the third portion of the MnSP and coincidence period.
It seems reasonable to assume that this spinal interneuron inhibition disorder may be associated with the occurrence of clinical events such as PKD, which could be triggered by an excessive synchronisation of the motor units in agonist and antagonist muscles.
However, further in-depth studies are required to clarify the mechanisms underlying PKD.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
We thank Meggie Chochol for editing the manuscript.

