All published articles of this journal are available on ScienceDirect.

Alzheimer Disease Pathology in Middle Age Reveals a Spatial-Temporal Disconnect Between Amyloid-β and Phosphorylated Tau
Authors Info & Affiliations
Abstract
We studied the brain distribution of amyloid-β (Aβ) and phosphorylated tau (τ) in 20 consecutive autopsy cases between the ages of 51 and 65, with no history of neurologic disease during life. We note that early accumulations of Aβ and τ occur in distinct neuroanatomical distributions. In the locus ceruleus and medial temporal lobe allocortex τ often occurs in the absence of diffuse Aβ and that Aβ occurs in the neocortex in the absence of τ. In those cases with both Aβ and τ were present in the sections, there was no overlap at the microanatomical or cellular level. APOE genotype was also assessed, showing no specific relationship with the presence or distribution of Aβ and τ, although the numbers of cases were limited. These findings indicate that the early appearances of hallmark proteins of Alzheimer’s disease are disconnected both in time and in space, suggesting that both are reactive phenomena with no mechanistic relationship in aging or preclinical disease.
INTRODUCTION
Neuropathological changes in the brains of the cognitively intact elderly have been the subject of numerous investigations [1]. While some marginal differences among studies have been reported, the data overall indicate that i) amyloid-β (Aβ) and phospho-tau (τ) pathology are both common in aged individuals absent cognitive dysfunction, ii) that Aβ pathology in the necortex and blood vessels may be substantial in this group of patients and iii) that τ pathology, while common particularly in the medial temporal lobe and brainstem [2], is rarely present in abundance in the neocortex (i.e. Braak stage VI) of the cognitively intact [3]. These data juxtaposed with the standard notion of the linear relationship between Aβ and τ per the amyloid cascade version of disease pathogenesis, has led to some controversy regarding a potential spatial and temporal disconnect in the biogenesis of fibrillary Aβ and insoluble τ [4], and by extension the applicability of the so-called Aβ and τ cascade hypotheses to the human brain in vivo.
We therefore sought to study Aβ and τ deposition in hospital autopsy cases of middle aged subjects in preclinical disease, such that we may gain insight into the temporal relationships with the confounding negative influence of advanced pathology. ApoE genotyping was also performed in a subset of cases.
MATERIALS AND METHODS
Fixation and processing
Brain tissue from 34 subjects was obtained at autopsy. All specimens were fixed by immersion in 20% neutral buffered formalin. After two weeks of fixation, the brains were examined externally, the cerebral hemispheres were sectioned in the coronal plane, the brainstem was sectioned in the axial plane, and the cerebellum was sectioned in the mid-sagittal and parasagittal plane. Standard brain sections were obtained and processed through graded ethanol and xylene solutions, embedded in paraffin, and sectioned at 8 microns. Sections containing pons with locus ceruleus, medial temporal lobe with hippocampal formation, and frontal premotor cortex (Brodmann area 8) were selected for immunohistochemical studies.
Immunohistochemistry
Deparaffinized sections were used for immunohisto-chemistry. Sections were deparaffinized through two changes of xylene, and then rehydrated through a graded ethanol series to Tris buffered saline (TBS; 50 mM Tris, 150 mM NaCl, pH = 7.6). Antibodies used were phospho-tau monoclonal antibody AT8 (Thermo Scientific), which recognizes pSer202/pThr205 of PHF tau, and monoclonal antibody 4G8 (Chemicon International), which recognizes an epitope between residues 18-22 of the beta amyloid peptide. For 4G8 immunohistochemistry, deparaffinized sections were placed in 88% formic acid for four minutes. For both 4G8 and AT8 immunohistochemistry, sections were placed in a pressure cooker at 126 psi for 20 minutes. Endogenous peroxidases were then abolished with a thirty-minute
incubation in 3% H2O2. After blocking sections with 10% normal goat serum (NGS) in TBS for thirty minutes, primary antibodies were applied for sixteen hours at 4 ºC. Sections were then rinsed in 1% NGS in TBS, followed by ten minutes in 10% NGS, and a species-specific secondary antibody made in goat was applied for thirty minutes. After rinsing again as previous, the species-specific peroxidase-anti-peroxidase complex was applied for one hour at room temperature. Sections were rinsed two times in Tris buffer and developed with 3-3’ diaminobenzidine (Dako). Stained sections were dehydrated and mounted with Permount.
ApoE genotyping
ApoE genotyping was performed on paraffin-embedded sections of cerebellum using the Promega Wizard Genomic DNA Purification Kit (#A1120). 10-20 mg of brain tissue was put in blue homogenizer tube (Fisher #K79520-0000), to which was added 600 µl of cold Nuclei Lysis Solution and homogenized for twenty seconds. The lysates were incubated at 65 °C for fifteen to thirty minutes. 3 µl Rnase Solution was added, and then incubated at 37 °C for fifteen to thirty minutes. The sample was cooled, and 200 µl Protein Precipitation Solution was added and subsequently vortexed. The samples were cooled on ice for five minutes, spun for four minutes (14000 rpm); the supernatant transferred to a clean tube and precipitated with 600 ul isopropanol at room temp. The white, threadlike DNA was aspirated, washed with 600 µl of 70% ethanol, vortexed, spun for one minute, and aspirated as before. The pellet was air-dried for ten to fifteen minutes, resuspended in 100 µl DNA Rehydration Solution, and incubated one hour at 65 °C.
PCR was performed using the Roche Expand High Fidelity PCR system using 5 µl DNA, 2mM MgCl2, 10% DMSO, 0.2 mM dNTP, 0.5 pmol APOE upstream (TCC AAG GAG CTG CAG GCG GCG CA), 0.5 pmol APOE downstream (ACA GAA TTC GCC CCG GCC TGG TAC ACT GCCA), and 1x buffer in a 50 µl reaction. After denaturing at 95 °C for five minutes, the thermocycler was set for 35 cycles (95 °C one minute, 65 °C one minute, 72 °C one minute). PCR products were precipitated and resuspended in water. Restrictions with CfoI (Promega) were performed for ninety minutes at 37 °C. Bands were separated on 8% acrylamide gels using TBE buffer.
Chart review
Following review with the Institutional review board, the study was exempted. Available clinical records were examined and summarized for all cases.
RESULTS
Routine pathological examination
Gross brain examination along with examination of representative sections stained with H&E showed no specific neuropathological changes. Isolated neurofibrillary degeneration was noted on routine stains in the entorhinal cortex and the locus ceruleus in a minority of cases.
T immunohistochemistry
Given the limited overall extent of proteinopathy in middle age versus advanced AD, cases were assessed as presence or absence of protein, rather than extent, which was limited in all cases when present.
T immunoreactivity was present in the locus ceruleus in all twenty cases. The immunoreactivity consistent of neuropil threads in all cases, as well as frank neurofibrillary tangles in a subset of cases. Similar findings were noted in 17 of twenty sections of the basal nucleus of Meynert, and in 19 of twenty sections of the medial temporal lobe allocortex including Ammon’s horn and the entorhinal cortex. Scattered neurofibrillary threads were detected in the frontal neocortex in 7 out of twenty cases (Figs. (1-4), Table 1).
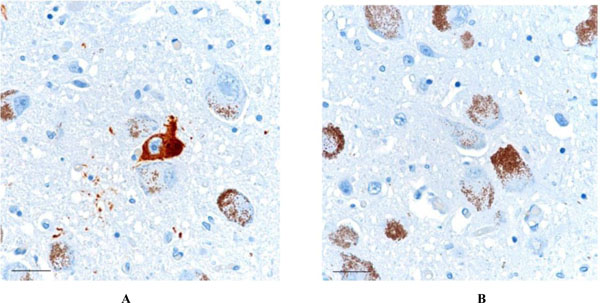
AT8-positive neurofibrillary tangle is noted in the locus ceruleus (1A - left). Some degree of τ pathology was present in the locus ceruleus in all subjects, in only one immunostain of a 6 micron thick section. No Aβ was present in the locus ceruleus region in any of the cases (1B - right). Scale bars = 30 μm.
Focally dense accumulation AT8-positive neurofibrillary tangles and neuropil threads is present in the CA-2 region of the hippocampus (2A - left), whereas no Aβ immunoreactivity was present, either in the CA-2 region or in the sections of cerebral cortex elsewhere in this case (2B - right). Scale bars = 100 μm
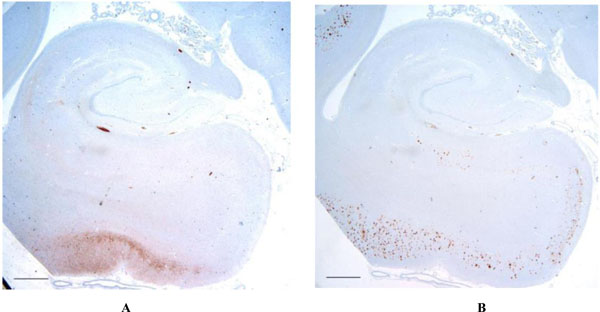
Low magnification of the hippocampal formation shows the distribution of t (3A - left) and Aβ (3B - right) in the case with the most advanced protein accumulation. Note that while some degree of overlap between τ and Aβ are present, significant areas such as in the region of the entorhinal cortex and subiculum, show Aβ in the absence of t. Scale bars = 1 mm.

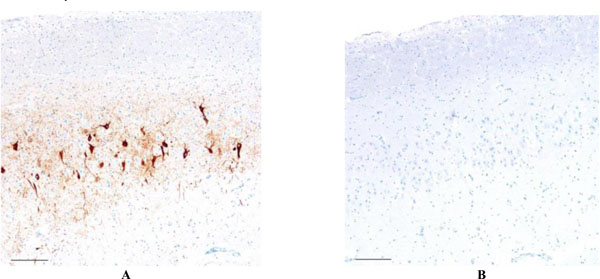
Focal AT8 positive neurofibrillary degeneration is noted in the frontal neocortex (4A - left), which was unaccompanied by Aβ pathology anywhere in the cerebral cortex (4B - right). Scale bars = 50 μm
Presence of τ and Aβ as a function of brain region and APOE.
| APOE | Tau | Amyloid-β | ||||
|---|---|---|---|---|---|---|
| ε2/E3 | Neocortical | 1/2 | Neocortical | 1/2 | ||
| Allocortical | 2/2 | Allocortical | 0/2 | |||
| Locus ceruleus | 2/2 | Locus ceruleus | 0/2 | |||
| NBM | 1/2 | NBM | 0/2 | |||
| E3/E3 | Neocortical | 4/14 | Neocortical | 8/14 | ||
| Allocortical | 13/14 | Allocortical | 6/14 | |||
| Locus ceruleus | 14/14 | Locus ceruleus | 0/14 | |||
| NBM | 12/14 | NBM | 0/14 | |||
| E3/E4 | Neocortical | 2/4 | Neocortical | 2/4 | ||
| Allocortical | 4/4 | Allocortical | 2/4 | |||
| Locus ceruleus | 4/4 | Locus ceruleus | 0/4 | |||
| NBM | 4/4 | NBM | 0/4 | |||
NBM = nucleus basalis of Meynert
Aβ immunohistochemistry
Diffuse plaques were identified in the cerebral neocortex in 12 out of twenty cases. The numbers varied from sparse to moderate. Neuritic and cored plaques were essentially absent. Medial temporal lobe allocortical involvement was present in 10 out of twenty cases, and generally to a lesser extent compared to neocortical involvement. The region of the locus ceruleus and nucleus basalis showed no Aβ deposits.
Combined presence of Aβ and τ
In five cases, both Aβ and τ were present in the cerebral cortex. However, in none of those cases were diffuse plaques and τ pathology colocalized, aside from one case in which both Aβ and τ were present in the inferior temporal neocortex, although the adjacent entorhinal cortex and subiculum showed τ in the absence of Aβ.
APOE genotype. PCR with restriction enzyme analysis demonstrated two ε2/ε3 subjects, four ε3/ε4 subjects, and fourteen ε3/ε3 subjects . These allelic frequencies are compatible with those found in the population. No specific τ or Aβ pathologies were noted as a function of APOE, although the numbers are too limited to draw definitive conclusions regarding the association of APOE with early τ or Aβ appearance.
DISCUSSION
The importance of studying middle aged subjects rests in the fact that Alzheimer disease pathology is end-stage essentially by definition, and like any other non-neoplastic condition, loses specificity the more the process is advanced [5]. Moreover, treatment efforts aimed at removing lesions or their constituent proteins, be they plaques or low-n soluble assembly intermediates, have failed in mild to moderate dementia [6], suggesting to some that intervention was too late and that earlier stages, such as mild cognitive impairment or even cognitively normal subjects, be studied alongside biomarkers. With this in mind, several factors with implications into Alzheimer disease pathogenesis emerged in this study, despite the relatively small numbers. First, accumulation of insoluble, phosphorylated τ, as evidence by AT8 expression, can and does occur, in the absence of Aβ, not only in the brainstem, but elsewhere in the brain as well. Indeed, in this study 20 out of 20 cases had evidence of the τ accumulation in the region of the locus ceruleus, while 0 out of 20 cases showed brainstem Aβ. Similarly, in the basal forebrain, 17 out of 20 cases showed τ deposits, and 0 out of 20 had Aβ. Neurofibrillary pathology appeared in the entorhinal region in 19 out of 20 cases, in which only 12 of these had diffuse Aβ deposits. In the frontal cortex, 11 out of 20 subjects demonstrated diffuse Aβ deposits, while 7 out of 20 subjects had τ deposits, typically in the form of neuropil threads. In those subjects who had both, the Aβ and the τ were not colocalized. Thus, it is clear that in this age range, the process of tau-phosphorylation, and the beginnings of neurofibrillary degeneration, either age related or Alzheimer disease related, precedes Aβ deposition in general, and Aβ specifically in the locus ceruleus by many years or decades [7].
APOE analysis showed two APOE ε2/ε3 subjects, four ε3/ε4 subjects, and fourteen ε3/ε3 subjects. These allelic frequencies are consistent with those found in the population. No differences in τ or Aβ pathology were noted as a function of APOE although the numbers more cases are needed to confirm the absence of an influence of APOE.
These findings also argue convincingly against a linear protein construct with Aβ in an upstream position and phospho-tau in a downstream, as it pertains to middle aged human brains in vivo. The evolving understanding of the neuropathology of aging is evident as well by the recent consensus guidelines for interpretation of AD changes at autopsy [8]. By these guidelines which is based on copious review of the literature, and the experience of numerous neuropathologists with expertise in AD, Aβ accumulation is considered to progress from neocortex, to allocortex, to deep gray matter, to cerebellum as disease advances. The reasons for this are obscure and nearly entirely uninvestigated when compared to basic neuroscience studies presuming AβPP as a neurotoxic starting point. There is further little evidence that the regional distribution of Aβ relates in any way to differences in regional or cellular APP transcription [9, 10]. Why some brain regions readily accumulate Aβ with age and disease, while other brain regions accumulate little to none, even with endstage disease, is unresolved.
In contrast to Aβ, the earliest accumulations of phospho-tau appear in the brainstem, followed by limbic structures, association cortices, and finally primary neocortical areas [2]. Moreover, phospho-tau generally appears in the brainstem prior to Aβ appearing in the neocortex. These simple data again defy the linear Aβ-phospho-tau construct, as regards the human brain in vivo. Similarly, the mechanistic basis for the early accumulation phospho-tau in some populations (e.g. locus ceruleus) and the late or absent accumulation phospho-tau in others, (e.g. cerebellar Purkinje cells), even with endstage disease, is unclear. These empirical data nevertheless demonstrate convincingly that there is a spatial and temporal disconnect between the accumulations of Aβ and phospho-tau, and that each can and does accumulate in the absence of the other [2, 4, 11]. Aβ and phospho-tau tend to converge with disease, but then only with advanced to endstage disease, and only above the tentorium for reasons that are obscure.
CONCLUSION
The results of this study indicate that Aβ and phospho-tau appear independently in middle aged brains and emphasize the complexity of the differences between human brain and theoretical protein cascades. It demonstrates convincingly that there is a spatial and temporal disconnect between the accumulations of Aβ and phospho-tau, and that each can and does accumulate in the absence of the other. Why some brain regions readily accumulate Aβ with age and disease, while other brain regions accumulate little to none, even with end stage disease, is unresolved and suggesting that both are reactive phenomena with no mechanistic relationship in aging or preclinical disease
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
None declared.

