All published articles of this journal are available on ScienceDirect.

An Immunological Analysis of Dystroglycan Subunits: Lessons Learned from a Small Cohort of Non-Congenital Dystrophic Patients
Authors Info & Affiliations
Abstract
The dystroglycan (DG) expression pattern can be altered in severe muscular dystrophies. In fact, some congenital muscular dystrophies (CMDs) and limb-girdle muscular dystrophies (LGMDs) are caused by point mutations identified in six glycosyltransferase genes which are likely to target different steps along the posttranslational “O-glycosylation route” leading to a fully decorated and functional α-DG subunit. Indeed, hypoglycosylation of α-DG is thought to represent a major pathological event, in that it could reduce the DG’s ability to bind the basement membrane components, thus leading to sarcolemmal instability and necrosis. In order to set up an efficient standard immunological protocol, taking advantage of a wide panel of antibodies, we have analyzed the two DG subunits in a small cohort of adult dystrophic patients, whom an extensive medical examination had already clinically classified as affected by LGMD (5), Miyoshi (1) or distal (1) myopathy. Immunofluorescence analysis of skeletal muscle tissue sections revealed a proper sarcolemmal localization of the DG subunits in all the patients analyzed. However, Western blot analysis of lectin enriched skeletal muscle samples revealed an abnormal glycosylation of α-DG in two patients. Our work reinforces the notion that a careful immunological and biochemical analysis of the two DG subunits should be always considered as a prerequisite for the identification of new putative cases of dystroglycanopathy.
INTRODUCTION
The dystrophin-glycoprotein complex (DGC) represents a group of cytoskeletal, transmembrane and peripheral proteins linking the extracellular matrix to the actincytoskeleton in a wide variety of human tissues, ranging from muscle (skeletal,cardiac or smooth) to central and peripheral nervous system, as well as the airways, lung or kidney epithelia [1]. Dystroglycan (DG) is the pivotal component of the DGC and is thought to play a crucial role in skeletal muscle, affecting both sarcolemmal stability and the post-synaptic elements at the neuromuscular junctions (MNJs) crucial for innervations [2]. The DG adhesion complex is composed of two subunits, α- and β-DG. α-DG is a membrane-associated extracellular matrix glycoprotein that binds with high affinity the extracellular matrix laminin and proteoglycans (perlecan, agrin and biglycan), whereas β-DG spans the membrane and links α-DG to the cytoskeleton via either dystrophin or utrophin [1].
Deletion of the DG gene in knockout mice resulted in early embryonic lethality [3] whilst DG expression could be “secondarily” altered in many other types of muscular dystrophy, including Duchenne muscular dystrophy [4, 5]. In particular, α-DG can lose its typical sarcolemmal targeting in dystrophin- and sarcoglycan-deficient muscle sarcolemmas, and DG expression was shown to be largely influenced in congenital muscular dystrophy type 1A [1, 6-8].
Recently, several forms of congenital muscular dystrophy have been linked to mutations in putative and demonstrated glycosyltransferases or accessory proteins of glycosyltransferases which are thought to be specifically involved in the posttranslational O-glycosylation pathway of α-DG [9]. In most cases, these disorders feature a marked alteration in the glycosylation pattern of α-DG and therefore are commonly referred to as secondary dystroglycanopathies [9]. To date, at least 8 genes have been linked to these disorders, namely the glycosyltransferases Protein-O-mannosyl transferase 1 (POMT1), Protein-O-mannosyl transferase 2 (POMT2) and Protein-O-mannose 1,2-N-acetylglucosaminyltransferase 1 (POMGnT1), three enzymes whose functions remain uncharacterized, Fukutin, Fukutin-related protein (FKRP) and LARGE, and two subunits (DPM1 and DPM2) of dolichol-phosphate-mannose (Dol-P-Man) synthase [9, 10] (see Table 1).
Congenital Muscular Dystrophies (CMD) and Limb-Girdle Muscular Dystrophies (LGMD) Characterized by Hypoglycosylation of α-DG
| Designation | OMIM | Gene | Protein |
|---|---|---|---|
| CMD* | |||
| MDC1C | 60661 | FKRP | Fukutin-related protein |
| MDC1D | 608840 | LARGE | Acetylglucosaminyltransferase-like protein |
| Fukuyama CMD | 253800 | FCMD | Fukutin |
| Muscle-Eye-Brain disease (MEB) | 253280 | POMGnT1 | O-linked mannose β1,2N-acetylglucosaminyltransferase |
| Walker-Warburg syndrome (WWS) | 236670 | POMT1 POMT2 |
O-mannosyl-transferase |
| LGMD | |||
| LGMD2I | 607155 | FKRP; DPM3 | Fukutin-related protein; Dolichyl-phosphate mannosyltransferase polypeptide 3 |
| LGMD2K | 609308 | POMT1 | O-mannosyl-transferase |
| LGMD2L | 611307 | FCMD | Fukutin |
| LGMD2M | 611588 | POMGnT1 | O-linked mannose β1,2N-acetylglucosaminyltransferase |
| LGMD2N | 613158 | POMT2 | O-mannosyl-transferase |
Adding to the complexity of dystroglycan-linked neuromuscular disorders, mutations in glycosyltransferase genes also cause muscular dystrophy displaying a later onset such as limb-girdle muscular dystrophies (LGMD) (Table 1) [9]. LGMDs are a group of clinically and genetically heterogeneous disorders characterized by weakness and wasting of pelvic and shoulder girdle musculatures. The clinical course of LMGDs varies from severe forms with early onset and rapid progression within the first decade of life, to milder forms with later onset and slower progression [11]. Several forms of LGMDs are due to mutations in members of the DGC, as sarcoglycans. Five forms of LGMDs (LGMD2I, LGMD2K, LGMD2L, LGMD2M, LGMD2N) are characterized by secondary hypoglycosylation of α-DG (Table 1) [12-16]. Also genetic abnormalities of dysferlin, a sarcolemmal protein involved in membrane repair, may cause both LGMD2B and Miyoshi distal myopathy [17]. It should be noted that no genetic abnormalities of the six genes encoding the O-glycosyltransferases have been linked so far to distal myopathies. Interestingly, the first human primary dystroglycanopathy associated to LGMD and cognitive impairment, featuring a DG “core protein” missense mutation (Thr192→Met) hitting the N-terminal portion of α-DG [18], has been very recently identified [19]. Following an extensive characterization, the authors suggest that the mutated site may influence the efficiency of binding and action of the LARGE glycosyltransferase, leading to a misdecorated α-DG [19]. Moreover, in a forward genetic approach aimed at finding novel genes involved in congenital muscle diseases, a zebrafish dystroglycan mutant hitting the C-terminal domain of α-DG was also identified, underlying the possibility of a primary involvement of dystroglycan in muscular dystrophies [20].
It is often quite difficult to distinguish between the various forms of LGMDs and distal myopathies. The diagnosis relies on a combination of immunohistochemical and immunoblot analysis followed by DNA sequencing for the identification of the primary gene involved. In this study we analyzed the DG subunits, α- and β-DG, in a small cohort of seven patients that have all been classified as affected by non-congenital muscular dystrophies by clinical examination, in order to assess the possible involvement of DG in LGMDs and distal myopathy phenotypes. Such an analysis can be particularly useful in order to pave the way for establishing a more standardized clinical and diagnostic assessment of the DG complex.
MATERIALS AND METHODOLOGY
Patients
Patients were diagnosed according to clinical observations, family history and serum creatine kinase (CK) levels. Muscle biopsies were obtained with informed consent.
Immunofluorescence of Skeletal Muscle Biopsies
Five unfixed serial cryosections, 10µm thick, were obtained from each muscle biopsy. After blocking of unspecific sites with blocking buffer (0.5% Triton X-100, 1% BSA in PBS) for 30 minutes at room temperature, the sections were incubated with the primary antibodies overnight at 4°C in a humid chamber. After washing in PBS, the sections were incubated with the appropriated fluorescent secondary antibody for 30 minutes at room temperature. All tissue sections were mounted in aqueous mountant and viewed with a fluorescence microscope (Nikon), images collected exploiting a digital Nikon camera (Coolpix 5400) using the software X-Pro.
Electrophoresis and Western Blot Analysis of Muscle Biopsies
Muscle biopsies from dystrophic patients and control sample were homogenized in lysis buffer (125mM Tris-HCl pH 7.4, 2mM EDTA, 1% SDS, 1% Triton X-100) containing protease inhibitors; supernatants (total protein extract) were collected upon centrifugation. Proteins were separated by SDS-PAGE and transferred to nitrocellulose (NT) membranes. After blocking with 5% dry milk Tris-buffered saline (TTBS; 150 mM NaCl, 10 mM Tris-HCl buffer pH 7.4 and 0.05% Tween 20), the NT membranes were incubated o/n at 4°C with primary antibody. After washing in TTBS and in TTBS containing 3% milk powder for 30 min, the blots were incubated for 1 h with anti-mouse or anti-rabbit antibodies horseradish peroxidase-conjugated (Pierce).
After washing in 3% milk powder TTBS and several washes in TTBS, blots were developed using enhanced chemiluminescence (ECL) (Pierce). The immuno-reacting bands were visualized by exposing the membranes to Kodak X-OMAT films. Glycoprotein enrichment was performed on total protein extracts. Briefly, supernatants were incubated with Wheat Germ Lectin (WGL) Sepharose 6MB (Amersham) overnight at 4°C. After extensive washing with washing buffer (WB) (50mM Tris-HCl pH 7.8, 500mM NaCl, 0.1% Triton X-100), bound glycoprotein were eluted in WB with 250mM N-acetylglucosamine and used in Western blot.
Antibodies
In Western blot and in immunofluorescence experiments, β-DG was detected with mouse monoclonal antibodies anti-β-DG 43DAG/8D5 (Novocastra) diluted 1:25 and anti-β-DG BD (Becton Dickinson) diluted 1:500. α-DG was detected with mouse monoclonal antibodies anti-α-DG clone VIA4-1 (lot 32685, Upstate Biotechnology) and clone IIH6 (lot 22991 Upstate Biotechnology) diluted 1:500 and 1:200, respectively. Polyclonal antibodies directed against the mouse α-DG C-terminal region previously characterized [21] were used at dilution 1:100.
RESULTS
Clinical Examination of Patients
Three male and four female patients affected by non-congenital muscle dystrophies were studied. Age at onset was on average 25 years (range 10-35 years), and the average age at biopsy 32 years (range 24-48 years). Three patients, P2, P4 and P7, reveal a positive family history; on detail, P4 and P7 have one brother affected respectively, whereas P2 has one brother and one sister affected. The creatin kinase level was analyzed and the values were within a range of 1 to 40 times the normal value. In particular, P1 underwent biopsy before showing increased levels of creatin kinase.
All patients had a normal respiratory and cardiac function, with the exception of P2 who was affected by a coronary artery disease.
Some patients were also genetically characterized. In particular, the calpain (CAPN3), dysferlin (DYSF), telethonin (TCAP), lamin (LMNA), γ-sarcoglyan (γ-SGC), fukutin, POMT1 and fukutin related protein (FKRP) genes were analyzed. Among the genes analyzed, only P3, P5 and P7 displayed mutations in the dysferlin gene. The clinical details and genetic analysis outcome are reported in Table 2.
Summary of Clinical and Genetic Features of Patients
| Patient/Sex | Age onset | Family hystory | CK level (times normal) | Age at biopsy | Respiratory function | Cardiac function | Physical examination (Clinical diagnosis) | Genetic Analysis |
|---|---|---|---|---|---|---|---|---|
| P1/M | 30 | No | 3-5x | 27 | N | N | Weakness of gastrocnemii and posterior thigh muscles (Distal myopathy) | CAPN3,FKRP DYSF, TCAP, LMNA: normal |
| P2/F | 10 | Yes | 1-1.5x | 48 | N | Coronary artery disease | Proximal lower limb weakness with quadriceps and calf hypertrophy. Scapular winging, mild facial and upper limb weakness (LGMD) | CAPN3, γ-SGC, FKRP, LMNA, Fukutin, POMT1: normal |
| P3/M | 35 | No | 15-40x | 24 | N | N | Severe distal lower limb weakness, mild lower limb weakness (Miyoshi myopathy) | Mutations in DYSF* |
| P4/M | 30-35 | Yes | 3-5x | 35 | N | N | Calf hypotrophy, scapular winging, upper girdle weakness. Pes cavus (LGMD) | Only the brother was analysed: CAPN3, FKRP, DYSF, TCAP, LMNA, FKTN, POMT1: normal |
| P5/F | 18 | No | 10-15x | 33 | N | N | Proximal and distal weakness in the four limbs with distal onset (LGMD) | Mutation in DYSF** |
| P6/F | NA | No | NA | 27 | N | N | NA (LGMD) | NA |
| P7/F | 25-30 | Yes | 10-40x | 30 | N | N | Weakness and calf hypotrophy, then proximal spreading (LGMD) | Mutations in DYSF*** |
N= normal.
NA= not available.
* mutations: exon 21 1966 A>G (K656Q) + exon 23 2220_2205delins T fs X17 (T734S).
** mutation: exon 50 5626 G>A (D1876N).
*** mutations: exon 27 2875 C>T (R959W).
Immunofluorescence Analysis of the Dystroglycan Complex
Immunofluorescence analysis of the DG subunits was performed on skeletal muscle cryosections of all patients, with the exception of P4, using a panel of antibodies hitting different epitopes within the α- and β-DG subunits, as depicted in Fig. (1). The β-DG subunit was stained with two different antibodies hitting the extracellular region (BD) and the cytoplasmic domain of the protein (43-DAG). The β-DG signal in all the samples analyzed was comparable with the control section, and showed normal sarcolemma expression (Fig. 2). Also the membrane localization of the α-subunit was normal when detected with a polyclonal antibody raised against the core protein of the C-terminal domain of α-DG: furthermore, the positive staining with an antibody raised against the carbohydrate molecules revealed the correct glycosylation of the α-subunit in all patients (Fig. 2).

Scheme of the DG subunits displaying the epitopes recognized by different antibodies.
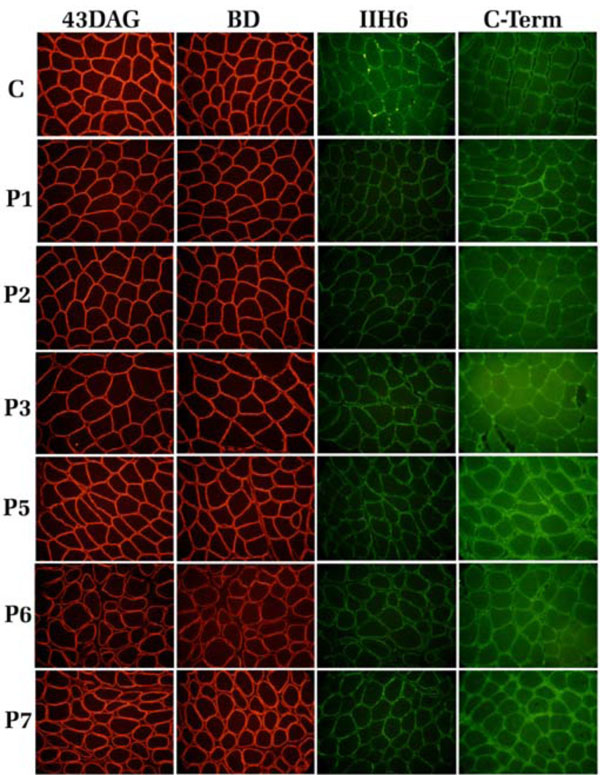
Immunofluorescence localization of α- and β-DG in human skeletal muscle sections. C: control, P1-P7: patients. The β-DG subunit was detected with two monoclonal antibodies, 43DAG/8D5 and BD, that recognized the β-DG cytoplasmic tail and its extracellular domain, respectively. α-DG was detected with the monoclonal antibody IIH6 (lot 22991), raised against an undefined carbohydrate epitope (red arrow), and with a polyclonal antibody directed against its C-terminal region.
Western Blot Analysis of Total Protein Extracts
The α- and β-DG subunits were also analyzed by Western blot using total skeletal muscle protein extracts. α-DG was probed with the monoclonal antibody VIA4-1 (Upstate), that recognises a yet unidentified carbohydrate epitope. The expected broad band of approximately 156 kDa was present in the control sample and in patients P1, P2, P3 and P7 (Fig. 3A, red arrow). The patient P5 displays a strong signal for α-DG but with a slight shift toward lower molecular mass (Fig. 3A). On the contrary, in patients P4 and P6 the 156 kDa band of α-DG was totally absent (Fig. 3A, red asterisk), although in P6 a very faint band around 120 kDa was also detected that could represent a hypoglycosylated form of α-DG still recognized by the antibody. β-DG was analyzed in total protein extracts using the monoclonal antibody NCL-43DAG that recognizes the last 15 aa of its cytoplasmic tail. All the samples analyzed showed the expected 43 kDa band (Fig. 3B).

Western blot analysis of the DG subunits in total protein extracts from patients P1-P7 and from control muscle tissue (C). A: Samples were resolved with a 7.5-15% acrylamide gradient SDSPAGE and α-DG was detected with the antibody VIA4-1 (lot 32685) raised against an undefined carbohydrate epitope (red arrow). In samples P4 and P6 the expected 156 KDa band was absent (red asterisk). The additional lower bands detected in all the samples might correspond to hypoglycosylated and/or proteolytic forms of α-DG; B: Western blot upon a 12% SDS-PAGE of the total protein extracts using anti 43DAG/8D5 for β-DG detection. All the samples showed the expected 43 kDa band.
Western Blot Analysis of Lectin-Enriched Extracts from Patients P4 and P6
The total protein extracts of patients P4 and P6, which showed an anomalous signal of α-DG in Western blot, were submitted to a lectin-enrichment protocol using wheat germ lectin (WGL) beads that specifically bind to N-acetylglucosamine residues. This protocol is very useful for the analysis of the carbohydrate decorations of a glycoprotein like α-DG. In the normal tissue sample, WGL-enriched fractions contain both α- and β-DG subunits (Fig. 4A). The WGL-enriched fractions from patients P4 and P6, analyzed with an antibody against β-DG, showed a loss of immunodetection in comparison to the corresponding total protein extract (Fig. 4B).
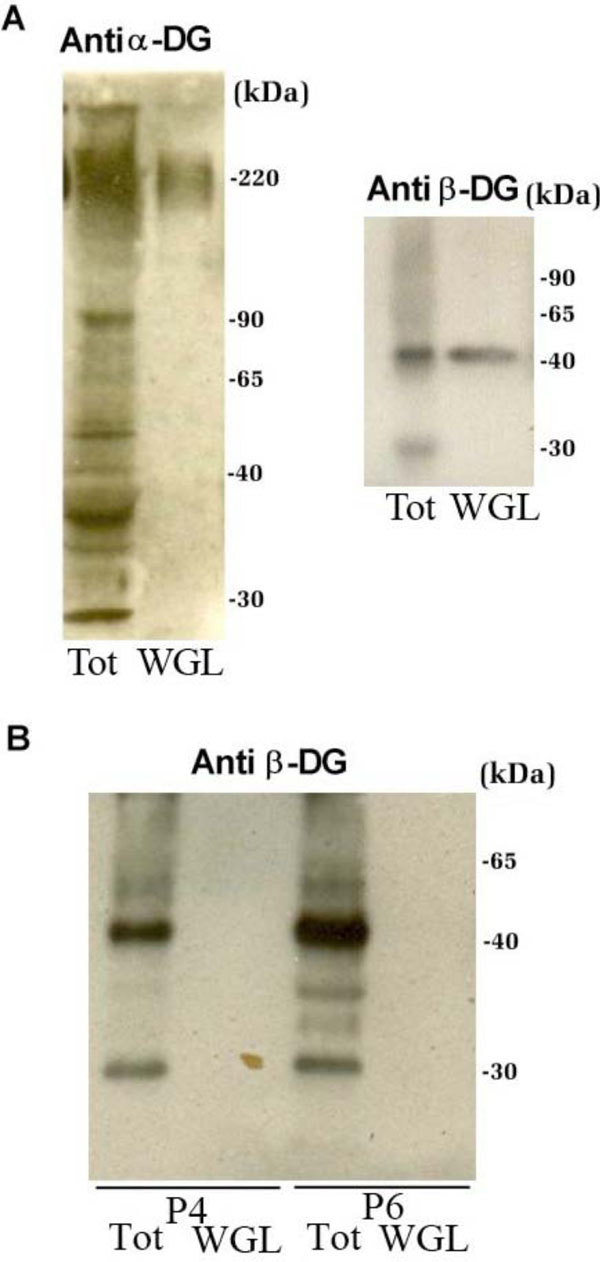
Western blot analysis of total protein extracts of control and patients P4 and P6 that were submitted to a lectin-enrichment protocol using wheat germ lectin beads. A: In the control tissue sample, WGL-enriched fractions contain both the α-DG and β-DG subunits. B: WGL-enriched fractions from patients P4 and P6, analysed with antibodies against β-DG. β-DG, that is present in the total protein extracts of both patients, is not enriched upon WGL incubation, confirming the presence of altered glycosylation of α-DG.
DISCUSSION
Primary and Secondary Dystroglycanopathies: An Increasingly Complex Scenario Ranging from Congenital to Limb-Girdle Muscular Dystrophies
In this study, we have analyzed the α- and β-DG subunits in seven patients who had been catalogued by clinical observation as subjects affected by non-congenital muscular dystrophies (Table 2). Five of those patients were classified as affected by LGMD, one by distal myopathy and another one by Miyoshi myopathy.
We have used a combination of immunofluorescence and immunoblot analysis to characterize the DG complex and to determine whether an alteration of the DG expression pattern emerged in any of these patients affected by non-congenital muscular dystrophies. Firstly, we analyzed the dystroglycan subunits expressed in skeletal muscle by immunofluorescence of tissue sections, using antibodies hitting different epitopes within the two molecules (Fig. 1). In particular, the β-DG subunit was stained using two monoclonal antibodies directed against its extracellular domain and its cytosolic C-terminal domain, respectively (Fig. 2), while the α-DG subunit was detected using a polyclonal antibody raised against the core protein at the level of its C-terminal domain, [21], and by a monoclonal antibody directed against an unknown carbohydrate epitope of α-DG (Mab IIH6 purchased from Upstate) (Fig. 2).
In all patients, all the different antibodies detected the expected membrane localization of the DG subunits (Fig. 2). From these very experiments, the DG complex does not seem to be related at all to the dystrophic phenotypes observed in those patients. However, Western blot analysis of crude muscle protein extracts, performed using a different monoclonal antibody directed against a carbohydrate epitope of α-DG (Mab VIA4-1, purchased from Upstate), showed the loss of α-DG immunodetection in patients P4 and P6, while patient P5 displayed a signal with a slight shift toward a lower molecular mass (Fig. 3A).
The discrepancy between the immunofluorescence and Western blot analysis may find an explanation in the possibility that α-DG glycosylation is indeed altered in these patients, and the carbohydrate epitope recognized by the VIA4-1 antibody is missing or is reduced. It should be noted that α-DG does not lose its sarcolemmal targeting, so it could be hypothesized that the altered glycosylation may rather affect its functional properties [22]. Such a picture seems to be confirmed by the lectin enrichment protocol that failed to co-purify β-DG from total protein extracts of P4 and P6 patients (Fig. 4B). In fact, the complete loss of immunodetection of the β-DG subunit in P4 and P6 WGL-enriched samples should depend on the impaired interaction between α-DG and the lectin, which in itself depends on the presence of N-acetylglucosamine residues in the mucin-like region of α-DG (Fig. 3A) [22].
Therefore, P4 and P6 patients may well be affected by a form of “dystroglycanopathy” that could be related to the reduced amount of N-acetylglucosamine present within the glycosylation shell of α-DG. Being POMGnT1 the glycosyltransferase that typically add the N-acetylglucosamine “brick” to the growing O-mannose sugar chains, these two patients (P4 and P6) could be tentatively catalogued as LGMD2M, even if muscle weakness displayed a distal involvement as well. Moreover, the patient P6 revealed also a deficit of calpain-3 (data not shown) that is characteristic of LGMD2A. Further genetic analysis will be essential to test this hypothesis and to identify directly and unambiguously the mutated gene responsible of the altered glycosylation of α-DG.
The Importance of an Extensive Immunological Analysis of the Two Dystroglycan Subunits for Clinical Diagnosis
Our analysis of a small cohort of patients revealed that the biochemical characterization of patients affected by muscular dystrophy should include a more extensive immunological analysis of muscle samples using different antibodies.
In fact, muscle sections of all the patients showed in immunofluorescence the same expression and targeting pattern of the two DG subunits. On the other hand, when applying Western blot analysis, especially upon lectin-enrichment, two patients were unambiguously identified as patients displaying an altered glycosylation pattern of α-DG. Although the possible role of POMGnT1 in a form of LGMD also with distal involvement could be only speculated, these patients should now undergo an extensive gene analysis aiming at a definitive characterization of the genes responsible for their pathologies.
After the identification of DG as a common final target for a whole series of glycosyltransferases responsible for its O-glycosylation pattern, the DG complex has taken centre stage in neuromuscular medicine [9]. Nonetheless, many important structural and functional details of the DG complex, in the light of its involvement in human neuromuscular disorders, await further elucidation. In particular, the novel identification of a pathogenic missense mutation in the DG gene that results in neuromuscular abnormalities, points to the opportunity of including DAG1 in the list of genes whose mutations cause muscular dystrophy and cognitive impairment in humans [19, 23].
Moreover, it has not been clarified yet whether DG would represent the substrate of other still unidentified enzymes. In fact, there are several reported cases in which patients, while displaying a series of phenotypic signs that would suggest the presence of a secondary dystroglycanopathy, do not carry mutations in any of the glycosyltransferases identified so far [9]. It is also possible that additional unknown substrates of these enzymes exist, enlarging the pathological scenario of sarcolemmal instability and necrosis [23].
During the next few years, further efforts will certainly be needed in order to identify alternative biochemical and/or genetic pathways underlying this group of congenital and limb-girdle muscular dystrophies.
ACKNOWLEDGEMENTS
This work was supported by Telethon grant GGP06225 to A.B. and Rare Diseases grant 7DR1 (ISS, collaborative project Italy-USA) to E.R. Association Française contre les Myopathies (AFM) is gratefully acknowledged for a post-doctoral fellowship to F.S.
The authors wish to thank Tamara C. Petrucci, Maria Giulia Bigotti, Enrico Bertini and Eugenio Mercuri (Roma) for critical reading of the manuscript.

