All published articles of this journal are available on ScienceDirect.

Pharmacological Treatment of Alzheimer’s Disease: Is it Progressing Adequately?
Abstract
Introduction:
Between 1993 and 2000 four acetylcholinesterase inhibitors were marketed as a symptomatic treatment for Alzheimer’s disease (AD), as well as memantine in 2003. Current research is focused on finding drugs that favorably modify the course of the disease. However, their entrance into the market does not seem to be imminent.
Research Development:
The aim of AD research is to find substances that inhibit certain elements of the AD pathogenic chain (beta- and gamma-secretase inhibitors, alpha-secretase stimulants, beta-amyloid aggregability reducers or disaggregation and elimination inductors, as well as tau-hyperphosphorylation, glutamate excitotoxicity, oxidative stress and mitochondrial damage reducers, among other action mechanisms). Demonstrating a disease’s retarding effect demands longer trials than those necessary to ascertain symptomatic improvement. Besides, a high number of patients (thousands of them) is necessary, all of which turns out to be difficult and costly. Furthermore, it would be necessary to count on diagnosis andprogression markers in the disease’s pre-clinical stage, markers for specific phenotypes, as well as high-selectivity molecules acting only where necessary. In order to compensate these difficulties, drugs acting on several defects of the pathogenic chain or showing both symptomatic and neuroprotective action simultaneously are being researched.
Conclusions:
There are multiple molecules used in research to modify AD progression. Although it turns out to be difficult to obtain drugs with sufficient efficacy so that their marketing is approved, if they were achieved they would lead to a reduction of AD prevalence.
INTRODUCTION
From 1906, when the first case of Alzheimer’s disease (AD) was described, until 1993, when tacrine was marketed, there were no specific treatments for this disorder. Between 1996 and 2000, new anticholinesterases entered the market and so did memantine in 2003. Currently, the marketing of new formulas for symptomatic treatment does not seem to be imminent and the interest of research is focused on finding products aimed at modifying the course of the disease. Unfortunately, years and years are going by and these drugs do not reach the market. This paper analyses synoptically the state of affairs of this research and the reasons hindering such an expected step in the history of AD.
SYMPTOMATIC TREATMENT
Since the 70s it is known that brain cholinergic deficiency is noticeable and early in AD, taking part in the genesis of certain manifestations such as hypomnesia. Tacrine (the first AD symptomatic treatment) is an acetylcholinesterase inhibitor (ACEI) which boosts brain cholinergic activity. It requires four doses a day and some patients do not have enough enzymatic resources in their liver to detoxify the products of its metabolism. New prescriptions of tacrine were interrupted when donepezil was approved in 1996, since the latter is well-tolerated and suitable for a daily dose. Subsequently, rivastigmine (1998) and galantamine (2000) were approved, two anticholinesterases which inhibit acetylcholinesterase to a lower extent than donepezil does. Rivastigmine compensates it through the simultaneous inhibition of butyrylcholinesterase and galantamine by exerting allosteric modulation of presynaptic muscarinic receptors, thus increasing acetylcholine secretion at the synaptic space. In clinical trials with ACEI against placebo, significant improvement was observed in cognition, behavior, functional autonomy and clinical global impression in patients ranging from incipient to moderately advanced AD [1]. A meta-analysis of 10 trials shows that, from 6 months onwards, patients treated with placebo worsen an average of 2.16 points on the cognitive scale ADASCog (70 points), while those treated with an ACEI in optimum doses undergo an average improvement of 0.27 points (average difference = 2.43 points) [1]. The difference among ACEIs’ effect does not exceed one point and thus meta-analyses do not find any significant difference regarding effectiveness on cognition among the three ACEI in use [1-3].
There exists a constant excess of glutamate in the synaptic clefts in diseased cells, which determines an excessive entrance of Ca++ into the cell through the existing channels in glutamate NMDA receptors. That’s why the post-synaptic cell is permanently depolarized, thus not being functional. Memantine, a NMDA receptor antagonist, prevents such continuous depolarization and keeps the usefulness of post-synaptic cells. Due to its voltage-dependent action mechanism low-moderate affinity towards NMDA receptors and a fast channel-blocking/unblocking kinetics, when a stimulus from the pre-synaptic cell arrives, it allows the opening of the Ca++ channels and a p action potential takes place in the post-synaptic neuron [4]. Thus, a population of neurons which had already lost their activity is able to prolong it due to the action of memantine. This explains its positive effect on cognition and functional autonomy [5, 6] as well as on behavior [7-9], allowing it to be marketed in 2003 for the treatment of patients in moderately advanced and advanced stages (with scores in the Folstein mini-mental state examination —MMSE— below 15). A meta-analysis of studies including patients with less advanced AD developed in 2007 allowed to extend its use to moderate AD (MMSE scores ranging between 15 and 19) [5, 10].
Although ACEI are more effective during initial stages of dementia and memantine in its advanced stages, both treatments are effective throughout the whole evolution of the Alzheimer’s disease. In fact, the US Food and Drug Administration (FDA) approved in 2007 the use of donepezil in advanced AD and, when experimentation with memantine in incipient AD prolongs, the positive effect obtained in the trials carried out so far could be verified [6, 11-13].
ADVANCES IN SYMPTOMATIC TREATMENT
Immediate marketing of new symptomatic treatments for AD does not seem foreseeable. However, the use of the ACEI available might be extended to the advanced stages of the disorder, following the path of donepezil. The appearance of new formats in 2007 and 2008 (galantamine in sustained-release capsules, donepezil in flas and rivastigmine patches) has increased the treatment’s tolerability, comfort and compliance. Memantine shows high tolerability and effectiveness in daily dose [14, 15], so that such a dosage has been accepted since 2008.
Some of the drugs researched to modify the course of AD have symptomatic effects (Table 1). If any of them were used as a progression modifier, it would also increase at the same time the arsenal of symptomatic treatments. It may also be that the symptomatic effect achieved will be precisely that which would allow them to enter the market.
Drugs with Symptomatic and Neuroprotective Action
| Action | |
|---|---|
| Dimebolin | ACEI + inhibitor of calcium L-channels and NMDA receptors |
| Huperzine A | ACEI + antioxidant and stimulant of muscarinic and nerve growth factor receptors |
| Phenserine | ACEI + sAPP-β and βA reducer |
| Memoquin | ACEI + I‑BACE, antioxidant and τ-hyperphosphorylation reducer |
| Bis-tacrine | tacrine dimmer. ACEI + I-BACE-1 and anti-NMDA |
| Lipocrine | tacrine (ACEI) and lipoic acid (antioxidant) hybrid |
| Tacrine-melatonin hybrids | ACEI + antioxidants |
| Ladostigil | rivastigmine (ACEI) and rasagiline (antioxidant) hybrid |
| Memantine | anti-NMDA + PP-2A stimulant (it decreases neurofibrillary degeneration), oxidative stress and activated microglia reducer |
sAPP-β: β variant of the soluble APP.
TREATMENTS THAT MODIFY THE EVOLUTION OF THE DISEASE
The knowledge of the etiopathogeny of AD is gradually increasing. A large number of mutations that lead to the development of familial AD, and numerous susceptibility polymorphisms that increase the risk of suffering sporadic AD, have already been identified (Table 2). An early and essential phenomenon is the formation of beta-amyloid (βA) and its aggregation, followed by a sequence of pathological events that lead to cell dysfunction and, subsequently, to premature cell death (Table 3). Certain susceptible brain regions are affected first, and the topographical expansion follows a regular chronology in typical cases [16, 17]. Researchers try to intervene in the steps of this pathogenic chain in order to slow down its morbid process. However, it is known that the administration of a neuroprotective drug does not necessarily involve any modification in the evolution of the disease and, when such a modification is achieved, it frequently does not take place to a significant degree.
Genes in which Determining Mutations and Susceptibility Polymorphisms (Associated to a Higher or Lower Risk) Related to AD have been Found
| Gene | Protein | Chromosome | Known FAD Pathogenic Mutations* | ||
|---|---|---|---|---|---|
| PSEN2 | presenilin 2 | 1 | 14 | ||
| PSEN1 | presenilin 1 | 14 | 173 | ||
| APP | βA precursor protein | 21 | 30 | ||
| SA** | ORa | ORb | |||
| APOE | apolipoprotein E | 19 | ε4 vs ε3 | 3.68 | 3.81 |
| CHRNB2 | β2 subunit of the neuronal nicotinic receptor | 1 | T vs G | 0.67 | 0.69 |
| GAB2 | GRB2 associated binding protein 2 | 11 | T vs G | 0.84 | 0.81 |
| CH25H | cholesterol 25‑hydroxylase | 10 | T vs C | 1.44 | 1.38 |
| SORL1 | Sortilin-related receptor | 11 | G vs C | 0.9 | 0.7 |
| CALHM1 | calcium homeostasis modulator 1 | 10 | T vs C | 1.42 | 1.42 |
| CST3 | cystatin C | 20 | C vs G | 1.23 | 1.28 |
| ACE | angiotensin I converting enzyme 1 | 17 | C vs T | 0.83 | 0.79 |
| PGBD1 | piggyBac transposable element derived 1 | 6 | A vs G | 1.25 | 1.25 |
| MAPT/STH | microtubule-associated protein τ/saitohin | 17 | T vs C | 1.24 | 1.24 |
* :Taken from http://www.molgen.ua.ac.be/ADMutations/ in November 2008.
** :Among the numerous genes in which susceptibility polymorphisms to develop sporadic AD have been found, we have pointed out those 10 genes which, up to now, have shown a greater degree of association (taken from http://www.alzforum.org/res/com/gen/alzgene/ in November 2008).
FAD: familial Alzheimer's disease.
a OR: Odds ratio obtained from the meta analysis of all available studies (a) and studies on Caucasians (b). SA: susceptibility alleles.
b OR: Odds ratio obtained from the meta analysis of all available studies (a) and studies on Caucasians (b). SA: susceptibility alleles.
Some of the Pathological Events that Take Place in AD
|
|
|
|
|
|
|
|
|
The symptomatic treatment acts by reducing a neurochemical imbalance which gives rise to particular symptoms (Fig. 1). When the treatment is withdrawn, its beneficial effect disappears (Fig. 2.4). When a drug improves the condition of AD patients, it is sometimes doubtful if the improvement is due to a symptomatic action or to a modification in the course of the disease, or if both effects act together [18]. There are some aspects of the evolution that are useful in checking if the course of the disease is changing [18-21]:
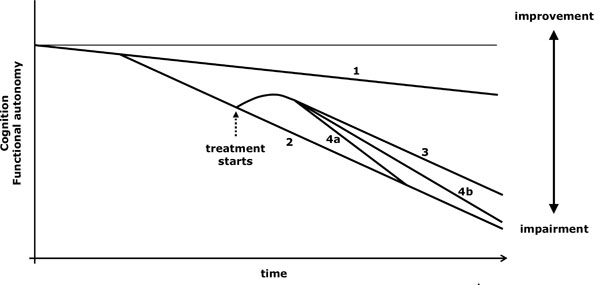
Virtual graph (*) of the possible effects of a symptomatic treatment. *: In reality, the natural evolution of impairment is not lineal and varies according to the stage of the disease; however, the lineal diagram facilitates the conceptual understanding of the action of the drugs. 1: Cognitive decline related to ageing. 2: Cognitive and functional loss due to an untreated progressive disease. 3: The optimum effect of a solely symptomatic treatment is maintained throughout. 4: Symptomatic treatment may produce a transitory beneficial action (4a) or a long-lasting benefit but to a progressively lower degree (4b).
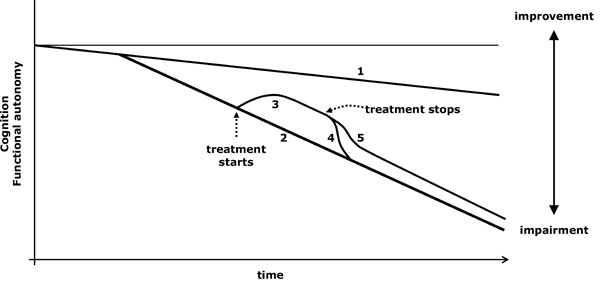
Virtual graph (*) of the possible effects of suspending a treatment with symptomatic effect. *: In reality, the natural evolution of impairment is not lineal and varies according to the stage of the disease; however, the lineal diagram facilitates the conceptual understanding of the action of the drugs.
1: Cognitive decline related to ageing. 2: Cognitive and functional loss due to an untreated progressive disease. 3: Improvement produced by treatment. 4: If a treatment is only symptomatic, its action is lost when it is suspended and the patient follows the evolution of untreated patients. 5: If the treatment, apart from being symptomatic, also modifies the course of the disease, its symptomatic action is lost when it is suspended, but the modification already achieved is held and the patient follows a progression line that does not overlap that of untreated patients.
- If a drug extends the time that elapses until a developmental moment is reached (a further stage of dementia, the need to be institutionalized, death, etc.), we may attribute evolution-modifying properties to it, once we have excluded the intervention of any other influent factor.
- A slower progression than expected also suggests that the drug is not merely symptomatic. In an evolutionary diagram, a progressively wider difference between the functional capacity of medicated and unmedicated patients would be observed (diverging lines) (Fig. 3.4 and 3.5). If progression speed is deducted from the slope of the evolution line, it is then necessary to compare the evolution of patients in equivalent clinical stages.
- When a solely symptomatic drug is stopped, the evolution curve will quickly overlap that of the untreated patients (Fig. 2.4). The improvement (or less impairment) achieved by an evolution-modifier drug does not disappear, or it does only partially, after being stopped (Fig. 2.5).
- Patients receiving a symptomatic treatment, once the necessary time to reach its maximum effect has gone by, should be comparable to other patients who are in the same stage of the disease and began the same treatment before. If the former are in a worse situation than the latter, it should then be inferred that the treatment modifies the course of the disease and, for such a reason, its early onset has long-lasting accumulative effects (Fig. 4).
- Paraclinical parameters acting as progression markers should show progression slowness in patients receiving drugs which modify the course of the disease. Magnetic resonance image of the hippocampal region, positron emission tomography (PET) with markers of amyloid plaques, or modifications of βA42 and tau or phospho-tau in cerebrospinal fluid are some useful elements for this purpose, although their validity has not been fully proven yet [20, 22]. Tests based on cerebral metabolism or perfusion (FDG-PET or HMPAO-SPECT, for instance) do not have the same validity, since some symptomatic treatments can modify them [18].
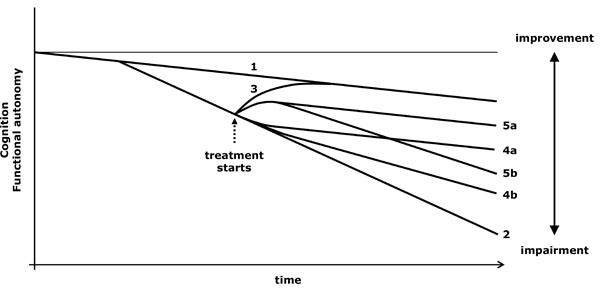
Virtual graph (*) of the possible effects of a treatment that modifies the evolution of the disease. *: In reality, the natural evolution of impairment is not lineal and varies according to the stage of the disease; however, the lineal diagram facilitates the conceptual understanding of the action of the drugs. 1: Cognitive decline related to ageing. 2: Cognitive and functional loss due to an untreated progressive disease. 3: Effect of a curative treatment (reversible disease). 4: Action of a treatment that modifies impairment progression speed, either by recovering the physiological slope (optimum situation, 4a) or by reducing progression speed (4b). 5: A treatment may have both symptomatic and modifying effects on the evolution of the disease, so that, after achieving an initial symptomatic improvement, the patient shows a diverging progression line in comparison to that of untreated patients, regaining (5a) or not (5b) the physiological slope (1).
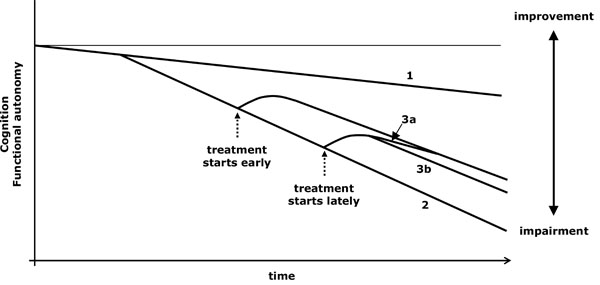
Virtual graph (*) of the possible effects of a symptomatic treatment. *: In reality, the natural evolution of impairment is not lineal and varies according to the stage of the disease; however, the lineal diagram facilitates the conceptual understanding of the action of the drugs. 1: Cognitive decline related to ageing. 2: Cognitive and functional loss due to an untreated progressive disease. 3a: If a symptomatic treatment starts late, the improvement catches that of patients who began treatment earlier. 3b: If the treatment also modifies the course of the disease, its late onset does not allow reaching the benefit obtained with an earlier onset.
I. Neuroprotective Action of Drugs Approved for the Symptomatic Treatment of AD
Several evidences point out that ACEI and memantine have a modifying effect on the evolution of AD. O. Lopez et al. have observed a higher interval until the patients administered ACEI are admitted to a nursing home [23] and, if memantine is added, such an interval is significantly higher and, moreover, survival time is also extended (paper by OL Lopez et al. read at the 60th Annual Meeting of the American Academy of Neurology, April 2008).
- Donepezil acts on the deep region of the acetylcholinesterase, where the acetylcholine is hydrolyzed, and on the surface area that interacts with βA and facilitates its aggregation. Its double nature gives it, apart from its symptomatic effect, a protective action against βA toxicity [24]. Other effects of donepezil, such as the stimulation of α-7 nicotinic receptors (which induces overexpression of the antiapoptotic bcl-2 protein) [25, 26] or the activation of σ-1 receptors (which reduces lipid peroxidation in hippocampal regions) [27], contribute to its potential neuroprotection. These effects may explain that hippocampal atrophy is slower in patients treated with donepezil [28, 29]. In a 3-year follow-up of two groups of patients (some treated with donepezil the whole time and others only during the last two years), patients treated with the drug since the beginning of the project retained better cognitive function at the end of the 3-year period (according to MMSE) [30]. The cumulative effect after a longer drug exposure represents a possible evolutionary modification (Fig. 4).
- The dual action of rivastigmine consists of inhibiting the hydrolysis of acetylcholine by acetylcholinesterase and butyrylcholinesterase. Since butyrylcholinesterase has greater impact on the formation of neuritic plaques than acetylcholinesterase [31], its inhibition may contribute to the lower cortical temporoparietal atrophy observed in patients treated with rivastigmine [32]. In a 5-year follow-up of patients treated with rivastigmine, MMSE scores show an increasing difference from the expected scores of untreated individuals [33] (model 5b in Fig. 3). The outcome should be interpreted cautiously due to the high number of patients lost during the follow-up period (basal sample: 1998, after 5 years: 83). In another study it was observed the evolution of patients who gave up the treatment prematurely, using three double-blind trials of rivastigmine against placebo; at the end of the 26th week, those patients who had taken rivastigmine in any previous period showed superior cognitive performance (ADAScog) than those of the placebo group [34]. This outcome, according to the graph in Fig. (2), points out that rivastigmine modifies the course of the disease.
- The stimulation of α-7 nicotinic receptors by galantamine enhances the expression of the antiapoptotic bcl-2 protein and has protective effects against glutamate excitotoxicity, stimulated by βA [25, 26, 35, 36]. In a 36-month follow-up of patients from two double-blind trials with galantamine against placebo of 6 and 3 months of duration, the scores on the cognitive scale ADAScog-11, which are always favorable to galantamine-treated patients, follow a diverging evolution in relation to the scores expected for untreated patients [37] (model 5b in Fig. 3).
- In many pathological states of the nervous system, AD among them, excitotoxicity contributes to functional and structural damage. In these situations, memantine experimentally shows a protective effect. This protective effect is reinforced by its ability to reduce the action of the activated microglia [38] and because it stimulates protein phosphatase 2A, thus reducing tau phosphorylation [39, 40]. After a 28-week double-blind trial of memantine against placebo, a 24-week open study was carried out. Patients treated with the drug continued with medication and those receiving placebo changed to memantine. In the end, the patients treated since the 29th week, in spite of having improved, did not reach the same beneficial situation regarding cognition and functional autonomy as those patients who had received memantine since the first day of the double-blind trial. The fact that no overlap took place between the curves may point out that memantine exerts a modifying effect on the evolution of AD [41] (see Fig. 4).
- Patients with probable AD treated with combination therapy (ACEI plus memantine) showed significantly lower mean annualized rates of deterioration in functional capacity when compared to patients who received ACEI alone or no treatment [42].
II. Drugs Under Research for Modifying the Evolution of AD
II.A. Drugs to Reduce βA Production
Insoluble βA is formed because γ-secretases (presenilins) and β-secretases (BACE -β-site APP cleaving enzyme-) cut the transmembrane amyloid precursor protein (APP), releasing this intermediate fragment, instead of being done by γ and α secretases, which release the soluble fragment APPs-α (Fig. 5). In the last few years, the strategy of inhibiting γ- or β-secretase has given a hint of hope in the treatment of AD. Most of γ-secretase inhibitors also inhibit the action of this enzyme on the notch protein and other protein substrates that take part in cell differentiation processes and therefore are not well tolerated (they produce alterations in thymus, intestine and spleen in experimental mice) [43-45]. For this reason, allosteric modulators or selective inhibitors of the γ-secretase which acts on the APP are currently being synthesized [45-47]. The γ-secretase inhibitor LY450139 has already started a phase-III trial. In the phase-II trial a tendency towards cognitive improvement and lower βA40 levels in blood and cerebrospinal fluid took place against placebo; however, no significant differences were reached [47]. The inhibitor may also cause eosinophilia and diarrhea and, in one case, death occurred after a digestive hemorrhage due to Barrett’s esophageal ulcer developed during the treatment [47]. Imatinib mesylate (antineoplastic) has shown an inhibiting action of γ-secretase without altering the notch substrate , so that its experimentation in AD has been suggested [48]. Several experts think that the action on β-secretase would be better tolerated, having tested BACE-1 inhibitors (I-BACE1) [49] and antibodies against the BACE cleavage site of the APP [50]. The GSK188909 I-BACE1, for instance, reduces βA load in transgenic mice [51], and CTS-21166 showed good tolerability and plasmatic reduction of βA in a phase I trial with healthy people. Successive generations of I-BACE have gradually obtained structures of low molecular weight and high inhibiting capacity for their possible use in clinical practice [52-60]. Apart from APP, BACE1 also hydrolyzes sialyltransferase ST6Gall (in the Golgi apparatus) and neuroregulin-1, which takes part in myelination processes occurring mainly after birth. Encouraging results have been obtained in BACE1 knockout mice, pointing out that the use of I-BACE in adults might not have negative repercussion [61, 62]. It is foreseeable that broad clinical trials with human patients will be undertaken soon. Among the multifunctional molecules under research, we can mention Memoquin which, apart from being I-BACE1, has antioxidant properties, is an ACEI and reduces βA aggregation and tau-hyperphos-phorylation [63].
Instead of inhibiting β- or γ-secretases, there is the possibility of stimulating the pathway of the α-secretase. Retinoic acid, final product of the metabolic cascade of vitamin A, modulates biological processes of proliferation, differentiation and apoptosis, stimulates the activity of α-secretase ADAM-10, protects from βA toxicity and is reduced in AD [64, 65]. Therefore, retinoic acid analogs such as fenretinide [65] or activators of its PAC1 receptor, such as the neuropeptide PACAP (pituitary adenylate cyclase-activating polypeptide) [66], constitute research elements to slow down the progression of AD. Among the multifunctional drugs which stimulate the non-amyloidogenic pathway of the α-secretase, statins and ladostigil can be included and shall be commented on later.
Some non-steroidal anti-inflammatory drugs (NSAID) such as ibuprofen, indomethacin, sulindac, diclofenac (and not others such as acetylsalicylic acid, naproxen, celecoxib and rofecoxib) have shown regulating effects on βA42 production. The mechanism for this action seems to be varied [67], but it mainly consists of the allosteric modulation of the γ-secretase that hydrolyzes the APP, without acting on notch or other substrates [68, 69]. Up to this point, no randomized prospective trials obtaining a significant favorable modification in the evolution of AD through NSAID treatment have been published. Glucocorticoids and celecoxib produce an amyloidogenic effect in some cases, so that it is not probable that their research in AD will continue [70-72]. In a broad epidemiological study a reduction of the risk of AD in individuals with the Apo E-ε4 allele who have been treated with NSAID was observed, but there were no differences according to the fact that the NSAID received was or was not a βA42-reducer [73-75]. On the other hand, knowing the side effects of NSAID (especially gastrointestinal effects), it should be proven that, after long-term administration, the potential modifying effect of AD is not cut short by serious complications. One of the γ-secretase modulating NSAID without action on notch is flurbiprofen. In the phase-II trial it was observed that in the subset of patients with incipient AD (MMSE = 20-26) and high dose of the drug (800 mg bid) significant improvement took place in functional autonomy and a positive tendency was also observed in cognition [76]. In the phase-III study, however, flurbiprofen failed to improve cognitive functioning or autonomy in daily activities.
Cholesterol increases βA-production from APP and, in turn, such increase reduces cholesterol synthesis. Statins inhibit lipidation of β-secretases and through the isoprenylation of GTPase, which takes part in secretases assembly to the APP, reduce βA-production [77, 78]. For this purpose, it is possible that the statins which cross the blood-brain barrier (simvastatin, lovastatin) are more effective than some others such as pravastatin or atorvastatin [79, 80]. They also have anti-inflammatory effects [81] and some of them, such as lovastatin, produce a glycogen-synthase-kinase 3 (GSK-3β) inhibitory effect [82], through which hyperphosphorylated tau protein is reduced and, in fact, less neurofibrillary tangles have been reported in patients who have previously taken statins [83]. The results from epidemiological studies and clinical trials regarding the risk to develop AD or modification of its evolution differ. Therefore, further evidence is needed before statins can be used as protectors against AD [84, 85]. The lipid-lowering drug fenofibrate, unlike statins, has an amyloidogenic effect similar to that of cholesterol [71, 72].
Apart from their sexual function, gonadal steroids exert other functions. Estrogens facilitate APP processing through the non-amyloidogenic pathway, reduce tau-hyperphosphorylation and oxidative stress, and exert a trophic stimulus on cholinergic neurons and other brain cells important in AD [86-91]. Epidemiological studies have observed lower incidence of AD in women who have received treatment with estrogens [92] and lower atrophy in hippocampal regions [93], although observational studies with negative results can also be found [94-96]. Among these, particular emphasis needs to be given to the Women’s Health Initiative Memory (WHIMS) [94], a randomized double-blind study which followed up (through an average of 4 years) 4532 nondemented postmenopausal women older than 64 years, who underwent treatment with conjugated equine estrogen and medroxyprogesterone (n=2229) or placebo (n=2303). Out of the 61 women who developed dementia, 40 (66 %) belonged to the group of active treatment and 21 (34 %) to the placebo group, which means a hazard ratio for probable dementia of 2.05, 45 vs 22 per 10000 person-years (p = 0.01). Participants came from a broader study (Women’s Health Initiative –WHI-) which was suspended in 2002 due to the incidence of adverse events. Discrepancy in the results of the studies is due to multiple reasons, although the main reason might be that estrogens slow the progression of AD only if they act over a prolonged period of time in an early pre-clinical stage, when there is still no significant βA accumulation. In fact, some researchers have observed that the administration of oestradiol to rats when βA42 has already been accumulated exacerbates neurodegeneration [97], and in short-term treatment [92] or in treatment of demented women [98] it no longer produces beneficial effects. In practice, we cannot prescribe this treatment in pre-clinical stages, due to the fact that we do not know markers for this stage. Besides, we should make use of a treatment whose long-term tolerability profile does not involve a high risk/benefit index. Emerging in this search for alternative drugs is the use of leuprorelin. AD patients show higher serum and brain levels of luteinizing hormone (LH), which facilitates APP processing through the amyloidogenic pathway [99]. Leuprorelin, analogous to the gonadotropin-releasing hormone (GnRH or LHRH), reduces LH and, in transgenic mice, gives rise to lower βA-production and cognitive improvement [100]. In September 2005 a phase-III trial began, whose results have not been published yet.
Phenserine is an ACEI that reduces APPs-β and βA production [101] (see Fig. 5). In a preliminary study with 20 patients, cognitive and cortical activity improvement (PET) was observed [102]. However, in two phase-III trials no significant cognitive benefit was obtained. In a post-hoc analysis of the three existing phase-III studies, cognitive improvement was observed in patients who received higher doses (15 mg/day) for more than 12 months, but no significant difference was reached against placebo regarding clinical global impression.
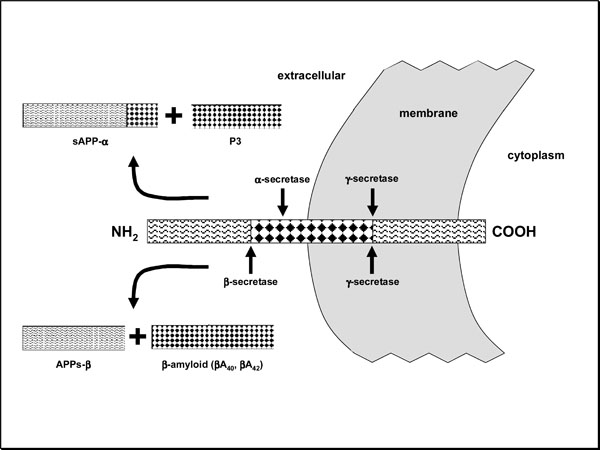
Transmembrane amyloid precursor protein (APP). In normal conditions, α- and γ-secretase act on it, thus producing the non-toxic fragments sAPP-α and P3. In AD β- and γ-secretase take part, releasing a less soluble fragment (βA) that is subsequently polymerized and forms insoluble deposits. This phenomenon is decisive for the disease’s pathogeny.
Cerebrolysine is a nootropic agent which reduces apoptosis and facilitates neurogenesis within the hippocampal region in transgenic mice [103]. Through a kinase-inhibiting effect (on GSK-3 and cdk5) it reduces phosphorylated APP, that gives rise to lower βA-production [104]. A meta-analysis of trials carried out so far points out that intravenous infusion 5 days a week for 4 weeks improves clinical global impression, although its effect on cognition and functional autonomy varies; in some studies it is no different than that obtained with placebo [105].
II.B. Drugs to Reduce βA Aggregation
There exist some synthetic peptides (beta-sheet breakers) —for instance, Aβ16-22m, iAβ5, iAb5p, iAb5p-A1, PAB-3631-PI, SEN-304— that attempt to prevent and undo the β-folding which takes place in many βA-fragments in order to reduce its aggregability and inhibit fibrillogenesis [106, 107]. Another strategy is the treatment with monoclonal antibodies aimed at the N-terminal region of βA involved in fibrils precipitation; it obtains fibrillar disaggregation, giving back solubility to the peptide and eliminating its toxicity [108]. All these proceedings still have not reached advanced stages in human experimentation.
Tramiprosate (Alzhemed®) is analogous to glycosaminoglycans which, through their joint to βA, hinders its aggregation. Unfortunately, in the last phase-III trial no significant clinical efficacy was reported and some researchers noticed that it may increase tau-aggregation [109]. Lipocrine, a hybrid compound of tacrine and lipoic acid, blocks the catalytic binding site of acetylcholinesterase and βA, giving rise to a reduction of βA aggregability as well as having antioxidant properties like the lipoic acid [110]. Colostrinin, derived from calostrum, solubilizes βA-fibres and reduces its polymerization, besides having antioxidant properties. In studies against placebo, it has been observed that it leads to certain cognitive and functional stabilization [111]. Curcumin, a component of culinary curry, crosses the blood-brain barrier and hinders βA aggregation; it is also disaggregating and has anti-inflammatory and antioxidant properties [112]. In a double-blind placebo-controlled trial with 34 patients, no significant clinical differences were observed and βA40 serum levels did not vary either [113].
Some metals take part in intracellular physiological oxidoreductive activity. Their interaction with βA has various effects. For instance, their bind to Cu++ (and Fe+++ to a lesser extent) facilitates the formation of toxic oligomers and the increase of oxidative activity and formation of free radicals, while the presence of Zn++ displaces Cu++ and exerts a protective effect [114]. Therapeutic strategies try to modulate the presence of these metals in the brain in order to reduce oxidative activity, or try to eliminate free radicals already formed by administering antioxidants. A zinc supplement in diet has been suggested, but experimentation points to its inadvisability [115]. Treatment with metal-chelating agents seems to be more plausible. Among the latter, clioquinol (PBT-1) did not obtain sufficiently positive result in the trial [116] and may produce visual alterations. PBT-2 is currently in phase-II of experimentation. It seeks to inhibit the formation of βA-oligomers and disaggregate amyloid plaques through promoting p copper and zinc brain homeostasis.
A phase-II trial with AZD-103 (scyllo-inositol, scyllo-cyclohexanehexol), which reduces βA aggregation and neutralizes oligomers, has already begun. NAP (also known as AL-108), administered intranasally, has also reached phase-II in research as an octapeptide with βA-antiaggregant and microtubule stabilizer properties. Memoquin has been mentioned in Section II.A as a multifunctional compound that reduces the βA aggregability promoted by acetylcholinesterase [63].
II.C. Drugs to Stimulate βA Elimination
A technique to eliminate βA is immunotherapy, which may be active (βA is administered to stimulate the production of antibodies) or passive (anti-βA antibodies are administered). In the first case we find AN1792, whose trial in humans was suspended due to the appearance of encephalitis. In the brain of autopsied patients who generated antibodies, lower than expected βA density was observed, apart from activated microglia and persistence of neurofibrillary tangles [117]. Clinically, no significant improvement against placebo was obtained [118] and greater loss of brain and hippocampal volume was observed through magnetic resonance [119]. The same dementia-progression rate was observed in patients showing βA reduction, perhaps because neuritic plaques are the final product of a neurotoxic process whose greatest responsibility may be attributed to amyloid oligomers, whose formation precedes plaque formation. On the other hand, a faster rate of progression was observed in patients with no amyloid reduction than in untreated patients, which adds fear to the use of this therapeutic technique. However, such line is still being researched; ACC 001 and CAD-106 (in phases II and I, respectively) are two formula of active immunotherapy that attempt to act without causing brain defensive reactions. Among monoclonal anti-βA antibodies we find AAB 001 (bapineuzumab). A study refers brain micro-hemorrhages in patients who underwent passive immunotherapy, so that further attention should be drawn to its tolerability [120]. The preliminary results of a phase-II double-blind trial with intravenous immunoglobulins against placebo in 24 AD patients for 6 months, showed better evolution in clinical global impression (p < 0.001) but no significant difference was reached regarding cognition (paper presented by N. Relkin et al. at the 60th Annual Meeting of the American Academy of Neurology, April 2008).
Insulin accelerates βA intracerebral circulation and elimination, and increases of the brain insulin resistance have been reported in many AD patients. Showing insulin resistance during midlife increases the risk of suffering AD in subsequent years [121]. For this reason, the effectiveness of rosiglitazone has been researched; it reduces such resistance, is anti-inflammatory and stimulates mitochondrial biogenesis [122]. In a double-blind placebo-controlled trial (n=518) cognitive improvement was only observed in the group of patients without the APO E ε4 allele [123] and, in another study in which 30 patients were followed up for 6 months, those patients who received rosiglitazone did not show the diminution of βA in cerebrospinal fluid shown by the placebo group [124]. It seems to be necessary to widen experimentation with this substance as well as to check to what extent it produces in AD patients the same cardiovascular events it causes in diabetes patients [125]. Intranasal insulin has also been researched since it increases glucose availability in the brain, scarcely affecting its peripheral metabolism. In a placebo-controlled study (n=25) treated patients showed better results regarding attention, memory and functional capacity [126]. As it occurs with rosiglitazone, it seems that the treatment is only effective in patients without the APO E ε4 allele [127].
Curcumin, already mentioned in the previous section, binds to amyloid deposits and facilitates their elimination [112], although such action produced no significant clinical or serum βA changes in a narrow trial [113].
Stimulation of plasminogen through tPA (tissue plasminogen activator) and the subsequent generation of plasmin reduces βA formation and accelerates its elimination. For this reason, the possible usefulness of a treatment with PAI-1 (PAI = plasminogen activator inhibitor) or TGB-β1 (PAI-1 inductor) inhibitors or antagonists has been raised [128]. It has also been observed, however, that tPA induces the activation of GSK3 and, thus, greater tau-phosphorylation and neurotoxicity [129], so that the application of the above mentioned treatment may turn out to be risky.
Somatostatin modulates βA-proteolysis mediated by neprilysin. In AD, a somatostatin depletion facilitates βA-accumulation. Drugs analogous to somatostatin (e.g. octreotide, ineffective in AD [130]), drugs which stimulate somatostatin release (for instance FK962, ineffective in a phase-II study) or drugs that act on its receptors, have been considered as candidates for experimentation aimed at improving the evolution of AD [131, 132].
II.D. Drugs to Reduce Neurofibrillary Degeneration
Phosphorylated tau-protein binds to microtubules in order to facilitate their assembly, assuring the stability of the neural cytoskeleton. A correct phosphorylation requires a balance between the action of some kinases and phosphatases (especially GSK 3, cyclin-dependent protein kinase —cdk5— and protein-phosphatase 2A —PP-2A—). GSK-3β plays a more relevant role than GSK-3α in tau-phospho-rylation, while the opposite occurs in APP processing to produce βA. There is a GSK3 hypothesis of AD, according to which GSK-3 hyperactivity would be responsible for excessive βA-production, periplaque microglia reaction and tau-hyperphosphorylation. The latter, in turn, alters axonal transport, reduces the firmness of the cytoskeleton and induces the formation of neurofibrillary tangles [133]. An increase of endogenous inhibitors of PP-2A in AD has also been observed [134]. From a therapeutic viewpoint, it may be possible to inhibit GSK-3 or stimulate PP-2A, having identified subgroups of AD that may respond differently to these treatments [135]. Molecules with anti-GSK3 properties have been investigated in antineoplastic research, some of them being suggested to be applied to AD, such as certain derivatives of indirubin [136], paullones [137], aloisines [138] or hymenialdisine [139]. Lithium and valproic acid have also been studied as GSK-3 inhibitors [140]. Valproate does not act on cortical GSK-3 [141] and, regarding lithium, some researchers think that it acts on GSK-3β and GSK-3α and, due to its action on the latter, reduces βA-production [142], while other researchers have observed βA-increases, which they attribute to a mechanism not related to GSK3 [143].
Among the multifunctional drugs with anti-GSK3 action, the afore-mentioned lovastatin [82] and thiadiazolidinones can be cited. The latter have anti-inflammatory and neuroprotective effect, partly because they activate nuclear peroxisome proliferator-activated receptors (PPAR) and partly because they inhibit GSK-3β [144, 145].
Nicotinamide (vitamin B3) produces cognitive improvement and phospho-tau reduction, mediated by its action on kinases, in transgenic mice (paper presented by KN Green et al. at the 60th Annual Meeting of the American Academy of Neurology, April 2008). Therefore, it is a candidate molecule to be valued in AD patients, maybe as a complement to other treatments.
On the other hand, memantine stimulates PP-2A, thus contributing to the reduction of neurofibrillary degeneration [39, 40].
Paclitaxel, which is related to taxol, stimulates tubulin polymerization and acts as a microtubule stabilizer [146].
Currently, active immunotherapy with phosphorylated tau is under research. The generation of antibodies that reduce neurofibrillary tangles with no inflammatory reaction has been observed in animals [147].
II.E. Drugs to Reduce Excitotoxicity and/or Oxidative Stress
Glutamate excess in synapses in diseased brain areas has deleterious effects for the cell (excitotoxicity). It causes excessive entrance of calcium into the cell, with an increase of the oxidative activity and functional damage to mitochondrias. LY451395, a modulator of glutamate AMPA receptors, did not reach significant clinical differences against placebo in a trial [148]. Dimebolin hydrochloride (Dimebon®) is an acetyl and butyrylcholinesterase inhibitor that blocks the entrance of calcium through L-type (voltage-dependent) and NMDA (glutamatergic) channels, having symptomatic and neuroprotective effects [149, 150], and modulating mitochondrial membrane pores, so that it helps preserve mitochondrial function [151]. In a one-year-long double blind, placebo-controlled, phase-II study with 183 patients, significant improvements in cognition, behavior and functional autonomy were observed [152]. Patients are currently being enrolled in a phase-III study (http://www.alzforum.org/new/detail.asp?id=1590;; http://www.medivation.com). Memantine is the antagonist drug of NMDA receptors that has given us the greatest amount of results within the field of AD. The reduction of excitotoxicity and of oxidative damage is one of its main functions [153]. Its proven symptomatic effect [5-10] and the fact of reducing tau-hyperphosphorylation [39, 40] make it an interesting drug with which to treat AD. Anyway, it will be necessary to prove its possible modifying effect in AD evolution through trials specifically designed for such aim. Other new-generation anti-NMDAs such as neramexane are currently being researched for neuroprotection in AD [154]. No significant differences were observed in a phase-III trial with 415 patients in which double-blind comparisons were established between a single anticholinesterase or a combination with neramexane; this research is ongoing with the administration of such drug as monotherapy against placebo.
Drugs with antioxidant action such as memoquin, lipocrine, colostrinin, curcumin, rosiglitazone, clioquinol or PBT-2 have already been mentioned in previous sections due to their multifunctional capacity. Some epidemiological and prospective studies observe lower AD incidence or progression in individuals taking vitamin E, preferably in combination with vitamin C [155-157], but a Cochrane analysis does not find clear evidence of any beneficial effect [158]. Furthermore, a meta-analysis showed an increase of mortality in individuals taking more than 150 UI/day of vitamin E [159]. Ginkgo biloba has antioxidant effects, but the most recent trials have failed to find any benefit on progression of cognitive impairment or AD [160-162]. Furthermore, such treatment is not free from a small risk of ischemic or hemorrhagic events [162, 163].
Melatonin has antioxidant and antiapoptotic properties, it increases mitochondrial energetic metabolism and reduces tau-hyperphosphorylation and βA-formation and deposit [164, 165]. Moreover, its MT1 and MT2 receptors are altered in AD [166]. Experimentation in AD animal models leads us to think of a favorable clinical effect of melatonin, but no controlled extensive clinical trial have been carried out in humans in order to demonstrate if melatonin can modify AD incidence or progression. Likewise, tacrine-melatonin hybrid molecules, which presumably would add the symptomatic effect of the anticholinesterase to the combination, are currently under research [167].
Studies developed with selegiline, an IMAO-β with antioxidant properties, conclude that there is no justification to use it in AD patients [168]. Rasagiline has different aspects from selegiline that make it more attractive, apart from facilitating APP non-amyloidogenic processing [169]. AD research, however, seems to have moved towards a bifunctional molecule, ladostigil, that combines properties of rivastigmine and rasagiline [169, 170].
Higher cortisolemia and greater cortisol/DHEA (DHEA = dehydroepiandrosterone, antiglucocorticoid) ratio are observed in the plasma of some AD patients than in healthy individuals of the same age [171], which may facilitate cognitive impairment and hippocampal atrophy [172]. For this reason, DHEA has been considered as a potential treatment of AD, since it reduces excitotoxicity and has antioxidant properties [173, 174]. In a narrow double-blind placebo-controlled study of DHEA, no significant differences were observed either in cognitive performance or in clinical global impression [175].
Huperzine A, an alkaloid of the herb huperzia serrata used in Chinese traditional medicine, has antioxidant properties, is an ACEI, stimulates muscarinic and neuronal growth factor receptors, increases APP non-amyloidogenic processing, protects against apoptosis and cytotoxicity caused by βA, glutamate or ischemia. In clinical trials carried out in China, it has been observed that it produces memory improvement in elderly people with amnesic disorder and in AD patients or those with vascular dementia, probably due to its ACEI action [176]. It would be desirable to see if its neuroprotective effects lead to favorable modifications in AD evolution. It is currently under a phase-II trial in USA.
A technique to increase the efficacy of already existing treatments is dimmer’s synthesis. Among them we can mention bis-tacrine (tacrine dimmer) and bis-HupA or bis-hupyridone (dimmer of a fraction of huperzine A). Bis-tacrine also has BACE-1 inhibitory action and blocks NMDA receptors, that provides it with an additional neuroprotective action [177].
Polyunsaturated fatty acids facilitate neuroplasticity in neuronal membranes and synaptic transmission, have antioxidant and antiapoptotic properties, protect against excitotoxicity, inhibit the production of proinflammatory cytokines and, furthermore, reduce βA and phospho-tau production while improving learning capacity [178-182]. These fatty acids are not synthesized in the brain but come from food. In animal models and in humans it has been observed that diets rich in these substances or containing a supplement of them (omega-3 fatty acids, docosahexaenoic acid) reduce AD risk, especially in individuals who do not carry the Apo E ε4 allele [183, 184], though some researchers have obtained negative results [185]. These products are available in the market as nutritional supplement. In a small group of AD patients a significantly favorable effect was observed in cognition (MMSE) from treatment with omega-3 fatty acids plus lipoic acid –antioxidant- (paper presented by L. Shinto et al. at the 60th Annual Meeting of the American Academy of Neurology, April 2008).
Propentofylline is a xanthinic derivative which inhibits adenosine reuptake and phosphodiesterase. Among other actions, it reduces the production of free radicals and microglia activation, stimulates the synthesis and release of the nerve growth factor, facilitates APP non-amyloidogenic processing and decreases activated GSK-3β [186]. The results from phase-II and III trials point out that improvement in cognition, global function and functional autonomy is achieved and the follow-up of individuals who left the study seems to indicate that propentofylline slows down AD progression [187, 188]. In a phase-II study it was observed that brain metabolism (PET) increased after stimulation with a verbal task [189]. However, the authors of the Cochrane analysis point out that part of the data obtained through experimentation has not been published by the researchers and that much of the published data does not discriminate between different etiological types of dementia [188]. The research of this drug in relation to AD has been interrupted.
II.F. Cell Therapy, Regenerative Medicine
The incorporation of multipotential cells into the brain to stimulate neurogenesis in brain areas affected by AD (cell therapy) is a promising field. In animal models, implanted cells undergo intracerebral migration, differentiate themselves and improve memory [190]. For multiple reasons, the treatment is complex. The optimum source of stem cells that should be implanted is a question currently under public and scientific debate. The implantation technique needs to minimize risks. The implanted cells should not only survive but they also need proliferate, differentiate themselves by acquiring the specialization of affected cells and, finally, integrate themselves in complex cell networks in order to make their function operative. The use of out-of-patient multipotential cells (obtained from embryonic tissues, bone marrow, umbilical cord or placenta, for example) may induce rejection more easily than using the patient’s own cells. In this sense, nerve cells have already been obtained from the patient’s skin or bone marrow [191]. Embryonic stem cells have great replication capacity but they also present a great risk for developing tumors [192]. An alternative consists of activating (through genetic modifications or nerve growth factors) stem cells which are inactive whether in the own brain or in another part of the body [191, 193, 194]. Moreover, this procedure avoids the surgery required to implant exogenous cells. A recent trial showed that stem cells administered intravenously to transgenic mice cross well the blood-brain barrier; this might solve the problems associated with implantation surgery [195]. On the other hand, if there is an excess of APP in the brain, multipotential cells become glial cells to a greater extent than neuronal cells (thus producing gliosis), so that it would be necessary to modulate the presence of APP. A variant of phenserine has been tested with such aim [196, 197]. Interesting to note is the study in which 6 AD patients were followed up for 22 months after being implanted genetically-modified autologous skin fibroblasts in order to produce human nerve growth factor. The results range from a slower progression of cognitive impairment to an increase of cerebral cortical activity (PET) [198].
Many of us, who are engaged in neurological clinical practice and are mere observers of this matter, suspect that other problems are much more difficult to solve:
- AD affects extensive associative and limbic areas of the brain as well as multiple subcortical gray-matter nuclei, which are the source of several neurotransmitters. Therefore, treatment with stem cells should act on various points and new cells should acquire varied specializations.
- When associative areas formed by axon and dendrite tangles of varied origin are affected, it seems difficult for newly arrived cells to properly integrate their new tentacles (both structurally and functionally) in such complex networks.
- Stem cells which begin to specialize within the diseased brain will have to do it in a hostile environment. When the first symptoms appear, several alterations —which include the activation of microglia and other inflammatory elements, accumulation of neuroexcitatory aminoacids, deposit of proteins with neurotoxic effects, etc.— have already been taking place in the brain for many years and many surviving cells are also functionally damaged, all of which hinders the development of implanted cells.
In short, it seems more feasible to introduce cells into the brain intending to stimulate the production of neuronal growth factors or the secretion of deficient neurotransmitters, instead of cells that attempt to substitute degenerated brain tissue.
On the other hand, knowing the possibility of creating transgenic mice, we may attempt to correct mutations and polymorphisms that cause the disease or increase the risk to suffer from it. This genetic manipulation is very difficult to apply in sporadic AD, since there are many genetic risk elements (many of which are still unknown) and difficulties of an ethical nature will be found in monogenic hereditary forms. In any case, what today seems to be utopian may become a pleasant reality within the not-too-distant future. However, the path currently followed by cell therapy and genetic manipulation and engineering to treat degenerative dementia may prove to be both thorny and lengthy.
REFLECTIONS ON THE DIFFICULTIES TO DEVELOP TREATMENTS THAT MODIFY THE EVOLUTION OF THE ALZHEIMER’S DISEASE
Historically, in 1976 the loss of cerebral cholinergic neurons in AD had already been detected [199], in 1979 an ACEI (physostigmine) had been tested [200, 201], in 1981 we knew the results of a study with tacrine [202] and in 1993 the same drug was approved by the FDA as a symptomatic treatment for AD. Technological and communication advances made us believe that the time intervals for approving new treatments would become progressively shorter. However, research on drugs which modify the course of the disease is considerably different from that which searches for symptomatic medication. For instance, in 1991 the relation between secretases and βA-synthesis was already known [203]. In 1999 it was established that presenilins are γ-secretases [204] and the characteristics of BACE (β-secretase) were detailed [205]. A year later the race had already begun to synthesize secretase inhibitors to treat AD. Unlike previous predictions it might have been foreseen, in 2008 there is still a long way to go to ready these molecules for market, if they do finally demonstrate their efficacy. As the first decade of the new century is coming to an end, none of the treatments aimed at modifying AD progression seems to be at the point of imminent approval, despite the hundreds of molecules now in different research stages, some of which are presented as effective in international meetings and congresses. What reasons can we give for our present situation?
Research on drugs that are directed toward brain neurotrophism, neuroprotection or neuroplasticity in AD patients, and that try to significantly reduce the progression of its preclinical process or clinical course, differs from the research that is necessary to prove the symptomatic effect of a drug [22]. Some difficulties are shared by all therapeutic research such as the possibility that effectiveness and tolerability (theoretically foreseeable or even observed in trials with animals) are not evident when experimenting with humans. When investigating drugs that modify the evolution of AD other difficulties are to be expected (Table 4).
Difficulties for the Development of Drugs that Modify the Evolution of AD
|
|
|
|
|
|
|
|
When the first symptoms appear in AD patients, the underlying neuropathological process has been brewing for decades [206]. If a drug could stop or slow down the physiopathological phenomena that provoke cell dysfunction and death in certain parts of the brain, it should be applied in preclinical stage with the aim of avoiding or delaying the symptom's appearance. Since AD is a disease that mainly appears at advanced ages, when mortality due to other causes is already very present, an extension of the preclinical stage would mean that many patients would not even show the first symptoms and some others would not go beyond the stage of mild cognitive impairment. That is, prevalence of dementia due to AD would be reduced. However, carrying out research on humans in the preclinical stage turns out to be extremely difficult. If we exclude known and asymptomatic carriers of a mutation producing familial AD, we do not have available suitable preclinical markers for the rest of the population. Besides, on the assumption that research took place in this group of pre-patients, the results could only be evaluated by observing the progression of histopathological lesions, since no clinical or biochemical markers -useful to create a base to see the effectiveness of the researched drug are known in such preclinical stages. Even if we assume that changes in the concentration of lesions are found (for instance, by using PET with specific markers such as Pittsburgh Compound B –PIB-) there would be little certainty that molecular and cellular pathology had been really delayed. It can be possible that βA-oligomers be harmful and plaques only constitute residues of an already-finished pathological phenomenon, so that a drug might reduce the concentration of amyloid plaques without obtaining any favorable clinical result.
When dementia is present it is less difficult to identify patients with probable AD. Neuropsychological markers, phospho-tau and βA42 alterations in cephalospinal fluid, hypoactivity of posterior cingulum extendable to other associative temporoparietal areas (through PET or SPECT) and noticeable atrophy in hippocampal regions (through magnetic resonance) allow practitioners to diagnose AD with high reliability, at least in typical cases without other alterations associated to it. However, AD is a heterogeneous disease. The genes that cause or facilitate its development are varied (Table 2) and, maybe for this reason, varied phenotypes are found in practice (frontal variant of AD, posterior cortical atrophy, AD starting with progressive aphasia or apraxia, AD with Lewy bodies). It is possible that, when researching drugs aimed at interfering in AD pathogenic sequence, it is valuable to know the patient’s phenotype supposing that, due to the disease’s heterogeneity, the effectiveness of the drug may differ within each subtype. Thus, it would be advisable to investigate within homogeneous, well-characterized groups. Regarding this aspect, we find lack of criteria and markers that identify AD phenotypes in a reliable way and, furthermore, we do not know in detail the existing differences among them at molecular and cellular level. If we accept simple clinical selection of phenotypes, the fact of working with subgroups would increase the difficulty of reaching sufficient sample sizes, which should be quite large in this kind of research, as it will be commented on later.
If drugs act by interfering in the activities of the diseased cells, in many cases they generate adverse effects, because they may also interfere with the activities of healthy cells or because they may bear unnecessary actions for the treatment of the disease. Such is the case of many gamma-secretase inhibitors, which not only reduce APP hydrolysis but also that of many other substrates, thus producing undesirable side effects. Another example can be found in memantine; due to its voltage-dependent and fast blocking-unblocking mechanism of action, it does not have problems related to tolerability, while other NMDA receptor antagonists previously researched (dizocilpine, phencyclidine, ketamine) produced severe adverse effects since they block these receptors in a more indiscriminate and permanent manner [4, 207]. Therefore, drugs characterized by high selectivity should be created in order to act only on something that works badly and only in ill cells. This process of increasing selectivity is time consuming and delays the availability of effective and well-tolerated drugs.
The expected effect of new treatments is the reduction of disease progression, although in many cases symptomatic improvement takes place simultaneously. In these situations, it is necessary to distinguish between both effects in order to demonstrate that the treatment modifies disease evolution. Techniques available for such an aim were described in a previous section of this article. Finding a difference becomes progressively more difficult as less time is available for observation. Regarding modification of disease course, there should be found not only statistical differences against placebo regarding clinical parameters, but also evidence of a measurable slowing effect (for instance, prolongation of the duration of dementia stages or the time of survival).
A low effectiveness of these drugs is possible, as they do not act on the whole etiopathogenic sequence, which means that it is going to be difficult to prove significant differences against patients who do not follow this treatment. If a drug were highly effective, it might be sufficient to show that the progression slope (throughout a period probably no shorter than 18 months) is significantly lower than that observed in the control group [19, 21]. However, in most cases it is probable that, in order to prove effectiveness, it is necessary to test the drug during several years in a large number of patients (several thousands of them, if we keep in mind the need to reach evidence as quickly as possible, obtaining non-intense benefit, and that a progressive loss of individuals is foreseeable throughout the follow-up process). The consequence of these requirements is a high cost that few companies are capable of assuming.
In order to lessen difficulties related to the scarce effectiveness of these drugs, the following strategies have been conceived:
- Searching for drugs which act on items close to the origin of the pathogenic cascade (e. g., β- or γ-secretase inhibitors), expecting to achieve greater effectiveness than that obtained with those which counteract an intermediate or final element of such a sequence (e. g., antioxidant substances or drugs which help eliminate amyloid plaques).
- Synthesizing products active in several aspects of pathogeny (examples shown in Table 5).
- Creating drugs with both neuroprotective and symptomatic effects (Table 1).
- Testing the effectiveness of cocktails of presumably-complementary substances. This option would turn out to be operatively more feasible if commercialized drugs, with already contrasted tolerability (see Table 5), were associated to one of the new molecules that have shown good tolerability and results, although not good enough for its approval.
Selection of Treatments Investigated to Modify the Course of AD. Multifunctional Drugs can be Found in More than One Section
| Action | |
|---|---|
| They reduce βA production | atorvastatin*a, bis‑tacrine, cerebrolysine*, fenretinide, flurbiprofen*, GSK 188909, huperzine Aa, ibuprofen*b, ladostigil, leuprorelin*b, LY450139b, memoquin, imatinib*, neuropeptide PACAP, simvastatin*b |
| They inhibit βA aggregation | AZD-103, β-sheet-brakers, colostrinin, curcumin, lipocrine, memoquin, PBT-2a, tramiprosate |
| They enhance βA elimination | PAI-1 or TGB-β1 antagonists, curcumin, active immunotherapy (ACC‑001, CAD‑106), passive immunotherapy (bapineuzumabb), intranasal insulin, rosiglitazone*b |
| They reduce neurofibrillary degeneration | aloisines, indirubin derivatives, hymenialdisine, anti-phospho-τ immunotherapy, lithium*, lovastatin*, memantine*, memoquin, nicotinamide*, paullones, thiadiazolidinones |
| They decrease excitotoxicity and oxidative stress | docosahexaenoic acid*b, ω3 fatty acids* + lipoic acid*, bis‑tacrine, colostrinin, curcumin, dihydroepiandrosterone*, dimebonb, tacrine-melatonin hybrids, huperzine Aa, ladostigil, lipocrine, melatonin*, memantine*, memoquin, neramexaneb, PBT‑2a, rosiglitazone*b, vitamins E+C* |
PAI = plasminogen activator inhibitor. TGB: transforming grow factor.
* : Currently marketed.
In phase-II (a) or phase-III (b) research, according to http://www.alzforum.org/drg/drc/default.asp [13.Nov.2008].
In another sense, the incorporation of pharmacogenomics into clinical practice within the next years is foreseeable. It may not be very useful in discovery new treatments, but it may help optimize the use of available drugs. Certain genotypes or polymorphisms condition qualitative aspects of the drugs’ target molecules or aspects related to their metabolism or other elements of the pathogenic chain, so that knowing them allows identifying subgroups of responders and/or non-responders in a certain treatment, or indicating those with good or bad tolerability. Let us consider some examples:
- Carriers of polymorphism rs733722 of the gene which codifies ChAT (choline-acetyltransferase) show greater response to treatment with ACEI [208].
- Patients whose GST gene (glutathione-S-transferase encoder) does not produce M1 or T1 isozymes suffer greater risk to develop hepatotoxicity if they take tacrine [209].
- Genotype CYP2D6 influences the plasmatic concentration and the effectiveness of donepezil [210]
- Patients without the K allele in the butyrylcholinesterase gene show greater response and less adverse effects when treated with rivastigmine [211].
- Certain alleles of pro-inflammatory genes may detect AD patients suitable to be treated with anti-inflammatories [212].
- Certain changes in the presenilin-1 encoding gene modify the antiamyloidogenic effect of anti-inflammatories. Mutations in exon-9 of PSEN1-δ remarkably reduce the efficacy of NSAIDs, while PSEN1-M146L and APP-V717F mutations increase their effect [213, 214].
- The PS1-L166P mutation induces ineffectiveness of LY-411575 (γ-secretase inhibitor) [214].
- In a study with rosiglitazone, favorable results were only achieved in patients without the allele APO E ε4 [123].
In summary, the race to develop treatments that modify the evolution of AD has obstacles and may produce a general impression of slowness, but it is unstoppable. The efforts carried out in order to solve the difficulties are huge and highly expensive, but they are not going to be in vain. Any drug that slows down the progression of AD will immediately have favorable repercussions on the disease’s prevalence and, in an ageing world, it will mean a great step in improving the prospects of health and wellbeing.

