All published articles of this journal are available on ScienceDirect.

Necrotizing Autoimmune Myositis: Anti-signal Recognition Particle and Anti-3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase Antibody Serotype Positive Patients with Varying Treatment Approach and Response
Dear Editor,
Necrotizing autoimmune myositis (NAM), also known as immune-mediated necrotizing, is a rare autoimmune myopathy and is often associated with statin use, inflammation, or malignancy. This is often correlated with autoantibodies anti-signal recognition particles and anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase anti- bodies [1].
Considering an interesting sero-association work by Ellis, Tan and Limaye, we present two NAM cases, the first report in our locale, documenting disease progression and therapeutic responses.
The first is the case of a 64-year-old woman with slowly progressive, proximal dominant, mainly lower limb weakness and dysphagia of over 7 months. Polymyositis being considered, she was initiated on intravenous pulse followed by oral methylprednisolone maintenance. While clinically improving over 6 months and reduction of total creatine kinase (CK: from initial 6,720 U/L to 131U/L), there was clinical recrudescence following use of pravastatin (shifted to simvastatin) for hyper- cholesterolemia. Total CK spiked to 5,607 U/L, prompting statin discontinuation and the addition of azathioprine. From then on, she was maintained on methylprednisolone and azathioprine combination that led to clinical improvement, yet with persistent mild weakness (MRC 4/5, proximally). An electrodiagnosis of myopathy prompted a muscle biopsy (Table 1) and eventually an autoantibody muscle panel that yielded a positive Anti-signal recognition particle antibody (anti-SRP). Rituximab was offered, but she chose to proceed with the same regimen plus rehabilitation. Since then, she has clinically stabilized with mild muscle weakness; she is independently able to do activities of daily living, but Total CK levels would fluctuate beyond normal values, although never above 1000 U/L.
The second is a case of a 50-year-old woman who initially presented with asymmetric lower limb weakness that became generalized weakness, which was associated with mild dysphagia over a 3-year period. Losing hope with clinical diagnosis of motor neuron disease, no treatment was taken. In the sixth year with the disease, she visited us, and a suspected chronic inflammatory myopathy was considered, given the course minus treatment and a slightly elevated total CK. At that time, she was in a wheelchair and had thigh atrophy, MRC 3/5 proximal-dominant symmetric limb muscle, and neck flexor weakness. From an electrodiagnosis of a chronic myopathy, a muscle biopsy was performed. Pulse intravenous followed by oral methylprednisolone was administered. After not having clinically improved further, a review of muscle biopsy findings (Table 1) led to performing an autoantibody muscle panel while adding azathioprine. Anti-3-hydro xy-3-methylglutaryl-coenzyme A reductase antibody (anti-HMGCR) was found positive. Subsequent sessions of intravenous immunoglobulin (IVIg) therapy yielded moderate clinical improvement (i.e., ambulatory with support); hence, rituximab was recommended.
NAM accounts for 19% of inflammatory myopathies. Just like other autoimmune conditions, and in the cases stated above, it has a female preponderance. Its usual age of onset is around the 5th decade of life [2]. Clinical features include subacute-progressive proximal muscle weakness as the hallmark [3]. Dysphagia may be present in 68% of anti-SRP and 44% in anti-HMGCR; respiratory insufficiency in 12% of anti-SRP and nil in anti-HMGCR; while skin and other muscular features are present in <10% of individuals [3,4]. These clinical features mirror that of the two cases presented above.
One-third (34%) of patients with NAM had documented evidence of statin use, which stands as the most common risk factor for developing NAM. Other risk factors include malignancy (predominantly gastrointestinal malignancies), inflammation, human immunodeficiency virus infection, and connective tissue disease. Hence, proper patient investigation for malignancy should be conducted [5,6].
Creatinine kinases are expected to elevate with a median peak CK of around 4700 IU/L in anti-HMGCR and anti-SRP myopathy. However, in some forms of myositis, CK levels may not accurately reflect disease activity. Additionally, some anti-HMGCR myopathy patients may masquerade as benign hyperCKemia [7]. In a chronic state, wherein there is significant muscle atrophy and fibrosis in biopsies, CK levels may be slightly elevated or normal, as seen in the second case [8].
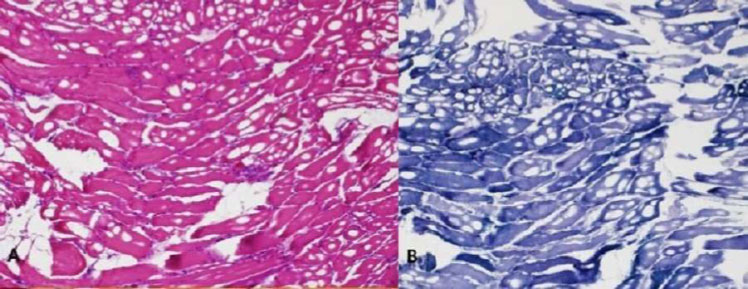
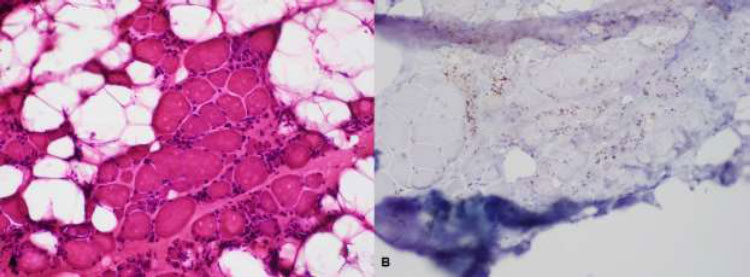
The majority of NAM patients respond well to immunosuppressive treatment; however, they may continue to progress despite intense efforts because of muscle inflammation that cannot be sufficiently controlled [9]. Most anti-SRP serotype-positive patients are resistant to steroid therapy and require combination and further aggressive treatment, as they tend to have more systemic manifestations, including interstitial lung diseases (Figs. 1 and 2) [3,4].
| CASE 1 | CASE 2 |
|---|---|
| 64-year-old, female, Filipino | 50-year-old, female, Filipino |
| Presentation: Insidious onset Proximal-dominant symmetric limb muscle, neck flexor and swallowing muscles affected over months. | Presentation: Initially asymmetric insidious onset then proximal-dominant limb muscle, neck flexor and swallowing muscles affected over years. |
|
Comorbidities: Diabetes mellitus Type 2 Dyslipidemia No skin problems nor malignancy |
Comorbidities: No skin problems nor malignancy No other comorbidity |
|
Initial Diagnosis: Polymyositis |
Initial Diagnosis: Motor neuron disease |
| Highest CK: 5,607 U/L (normal value: 30-145 U/L) | Highest CK: 750 U/L (normal value (30-145 U/L) |
|
Electrodiagnosis: Proximal muscles: brief small amplitude motor unit action potentials, with fibrillations; early recruitment; no fasciculations, myokymia and myotonia; with normal motor and sensory nerve conduction studies |
Electrodiagnosis: Proximal muscles: Brief small amplitude motor unit action potentials, with fibrillations; early recruitment; no fasciculations, myokymia and myotonia; with normal motor and sensory nerve conduction studies |
| Biceps brachii biopsy: Degenerating and necrotic fibers with macrophage invasion; few inflammatory mononuclear endomyseal infiltrates; no perifascicular atrophy nor perivascular and perimyseal inflammatory cells; no rimmed vacuoles; moderate increase in fibrous connective tissues. | Biceps brachii biopsy: Degenerating and necrotic fibers with macrophage invasion; few inflammatory mononuclear endomyseal infiltrates; no perifascicular atrophy nor perivascular and perimyseal inflammatory cells; no rimmed vacuoles; increased fibrous connective tissues and fatty replacement. |
| Autoantibody: Anti-signal recognition particle antibody (anti-SRP) positive | Autoantibody: Anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibody (anti-HMGCR) positive |
|
Final Diagnosis: Necrotizing autoimmune myositis (anti-SRP) positive |
Final Diagnosis: Necrotizing autoimmune myositis (anti-HMGCR positive) |
|
Treatment/s: Pulse IV methylprednisolone (1g/day for 5 days) Followed by maintenance regimen of oral methylprednisolone 8mg OD and azathioprine 100mg/day |
Treatment/s: Pulse IV methylprednisolone (1g/day for 5 days) Followed by maintenance regimen of oral methylprednisolone 8mg OD and azathioprine 100mg/day Subsequently on IVIg 2g/kg followed by 1g/kg every 3 months. |
|
Response: Moderate clinical improvement, ambulatory with support |
Response: Moderate clinical improvement, ambulatory with support |
CONCLUSION
In conclusion, further treatment with rituximab, as was recommended in these present cases, may be considered, especially for those with anti-SRP serotype resistant to steroids and other immunotherapies. Furthermore, IVIg might prove to be beneficial in patients positive with anti-HMGCR serotype who are refractory to or with contraindication to steroid therapy [10].
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

