All published articles of this journal are available on ScienceDirect.

Involvement of the Nervous System in Systemic Sclerosis
Authors Info & Affiliations
Abstract
Introduction:
Scleroderma is a rare heterogeneous multisystem autoimmune disease. The disease is characterized by structural abnormalities of the small blood vessels. Scleroderma affects all organs of the body. Skin manifestations are commonly reported, but peripheral nervous system (PNS) and central nervous system (CNS) involvement has been less frequently reported. Neurological manifestations are broad, and it is challenging for clinicians to confirm a diagnosis of scleroderma.
Case Presentation:
In our case, a 53-year-old white woman had extensive clinical presentations: skin rashes and symptoms from internal organs, CNS, and PNS during the previous 11 years. She had not undergone any specific treatment because diagnosis has not been made in the early stages.
Conclusion:
It is important to make the diagnosis as early as possible and start treatment with immunomodulatory and immunosuppressant medication, as it affects the patient's disease progression, quality of life, and mortality. A detailed medical history, physical examination, and laboratory and radiology findings help us to identify and diagnose scleroderma. But unfortunately, it was late, and the patient died. This case guides us to be more careful and make the diagnosis of scleroderma earlier in the future.
1. INTRODUCTION
Scleroderma is a rare, progressive, heterogeneous multisystem autoimmune connective tissue disease of unknown etiology, the pathogenesis of which is still not fully understood [1, 2]. The exact prevalence of systemic sclerosis is unknown because it is frequently misdiagnosed, and the data collected have been poor over the past years. Approximately 1 in 6500 adults are affected by it, predominantly women (4:1), and the age of onset is around 45 years [3, 4]. Scleroderma is characterized by small blood vessel vasculopathy, autoantibody production, and fibroblast dysfunction, leading to increased deposition of extracellular matrix [5]. Scleroderma is generally divided into two main forms: localized scleroderma and systemic sclerosis (or systemic scleroderma) [1], with visceral organs being affected. Systemic sclerosis can be subtyped into limited cutaneous, diffuse cutaneous, and systemic sclerosis without skin involvement [5]. Frequently, skin, lungs, gastrointestinal tract, kidneys, and heart are affected [2, 6]. It can manifest as thickening of the skin, telangiectasia, fingertip ulceration, Raynaud's phenomenon, interstitial lung disease, pulmonary arterial hypertension, gastroesophageal reflux, constipation, bacterial overgrowth of the small intestine, bloating, renal crisis, diastolic or systolic heart failure, arrhythmia, or conduction disturbances [5, 7]. Neurological symptoms are rare. Although the autonomic, peripheral, and central nervous systems are involved, the more common symptom expression comes from the peripheral nervous system [1, 6, 8]. There is no laboratory test or gold standard method for diagnosing scleroderma [9]. However, there are some criteria that should be used for diagnosis in clinical practice. Systemic sclerosis has a high mortality rate, which is why rapid recognition of the disease and early treatment are crucial to ensure the quality of life and improve survival for patients [4, 7]. Early initiation of therapy with an immunosuppressive agent, such as methotrexate, mycophenolate mofetil, cyclophosphamide rituximab, is highly recommended for diffuse cutaneous systemic scleroderma. In addition, oral steroids and azathioprine can be considered [7].
Central nervous system involvement is very rare. In some studies, white matter hyperintensities have been detected on magnetic resonance imaging (MRI) of asymptomatic patients. MRI changes do not depend on the duration or severity of the disease [1, 4, 10]. There are only a few cases reporting central nervous system involvement. We describe a case of systemic sclerosis in a 53-year-old woman with manifestations in the internal organs and nervous system. Scleroderma is a rare disease that is more associated with rheumatology and not neurology. This case was the first in our practice where neurologic symptoms were one of the first expressions, and the diagnosis was challenging.
2. CASE PRESENTATION
A 53-year-old woman with progressive asymmetric tetraparesis was hospitalized in the neurology department. Weakness in both arms and legs had been progressing over the previous year, and for this reason, the patient was using a wheelchair. She had ischemic neuropathy of the right optic nerve for 3 years. She had a separate pallid rash and thinner skin on her body for the past 11 years. From the beginning, there was a rash on her hands and the front wall of the abdomen, which gradually grew and appeared all over her body. Moreover, for 7 years, the patient had low blood pressure with orthostatic hypotension, and the previous year, she was diagnosed with sick sinus syndrome, for which a pacemaker was implanted. The patient had diffuse abdominal pain, gastroesophageal reflux, and constipation for a few years. She also had pain in the joints, which got worse in the mornings. The patient did not have any other chronic diseases.
Neurological examination at the time of admission revealed severely decreased vision in the right eye, but she could still distinguish light from darkness. In both eyes, she had impairment of horizontal eye movement and convergence. She had asymmetric tetraparesis with arm strength of grade 3 proximally and grade 3 distally; strength in the right leg was grade 0, and in the left leg was grade 4 proximally and grade 5 distally (scored according to the Medical Research Council scale). She was using a wheelchair, but she was still able to move from the wheelchair to the bed and back by herself and sit without assistance for a short time. Deep tendon reflexes were 2+ right > left. Babinski's sign was present on the right side. She had tenderness at the Th1-Th2 and Th11-Th12 levels. Vibration sense was 5/10 in both legs and 10/10 in the arms. She had urinary retention as well.
During skin inspection, a diffuse hypopigmented rash on her body was discovered, more over the anterior chest and abdominal wall, neck, hands, forearms, and legs, and telangiectasia on the anterior chest wall. Raynaud's phenomenon and puffy fingers were present. During the conversation, she had shortness of breath.
Extensive laboratory evaluation findings were normal, including full blood screening; erythrocyte sedimentation rate; C-reactive protein; coagulogram panel; thyroid function; and levels of creatine kinase, aminotransferases, electrolytes such as potassium, sodium, and chloride, glucose, vitamin B12, and creatinine. HIV antigen and antibody, anti-HB, anti-HCV, and rapid plasma reagin tests were all negative. Onconeural antibody panel, aquaporin 4 antibodies, and Fabry disease were also negative. Serum protein electrophoresis was normal. Immunological tests were normal, i.e., anti-C1Q antibodies, CD3, CD4, CD8, CD4/CD8, serum amyloid A, extractable nuclear antigen, anti-neutrophil cytoplasmic antibodies, anti-double-stranded DNA antibody, antiphospholipid antibodies IgM and IgG, and anti-cardiolipin antibodies C3, C4 and IgG4. However, beta-2 microglobulin was elevated (2.75 mg/L; normal range 1.01–1.73 mg/L), and antinuclear antibody was positive. Positive IgG Western blot for B. burgdorferi was also found. Urinalysis was normal. Blood and urine cultures were without pathological changes. Cerebrospinal fluid examination showed a slightly elevated protein level (0.57 g/L), and cell count was normal. IgG and IgM antibodies for B. burgdoreferi (by ELISA), Enterovirus (by RNA PCR), Varicella zoster (by DNA PCR), and Cytomegalovirus (by DNA PCR) were not present in cerebrospinal fluid. Oligoclonal bands were negative.
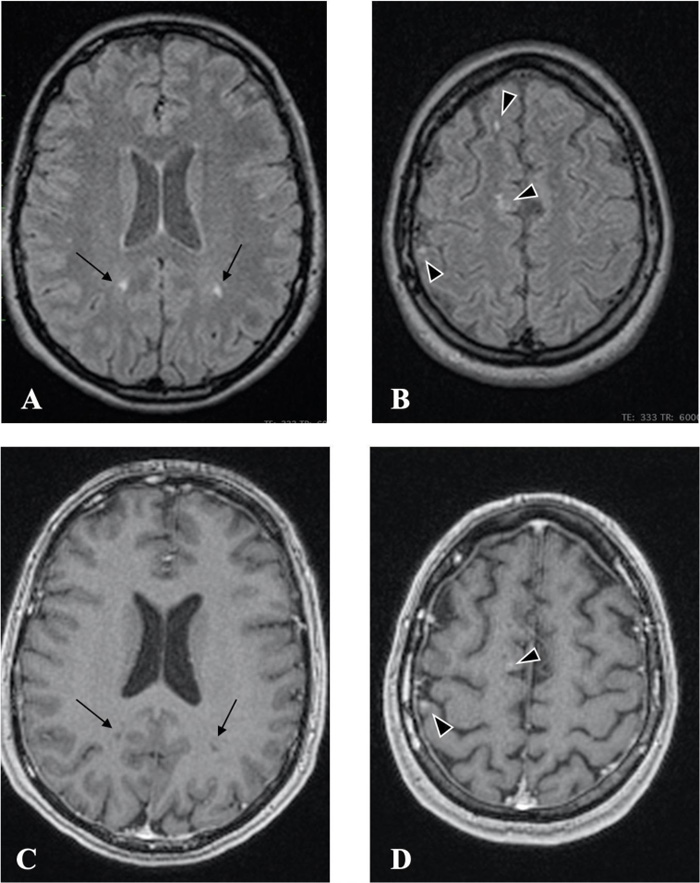
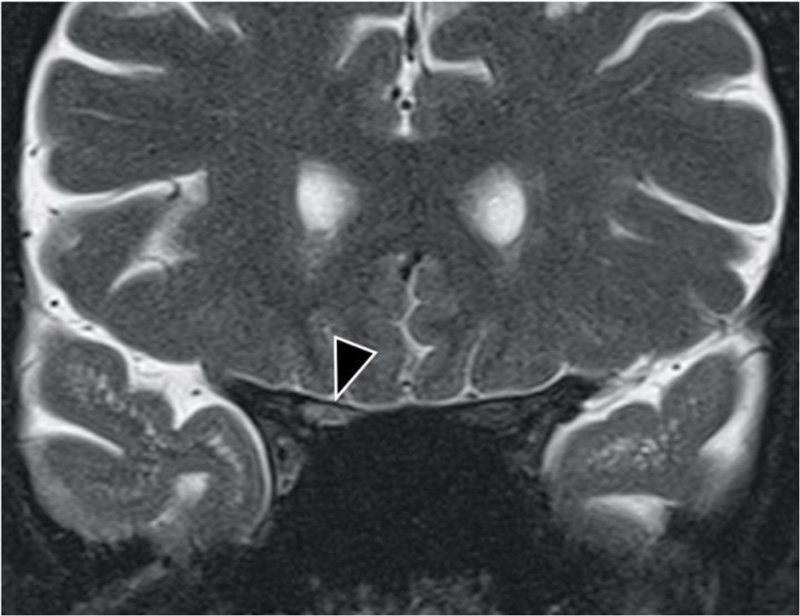
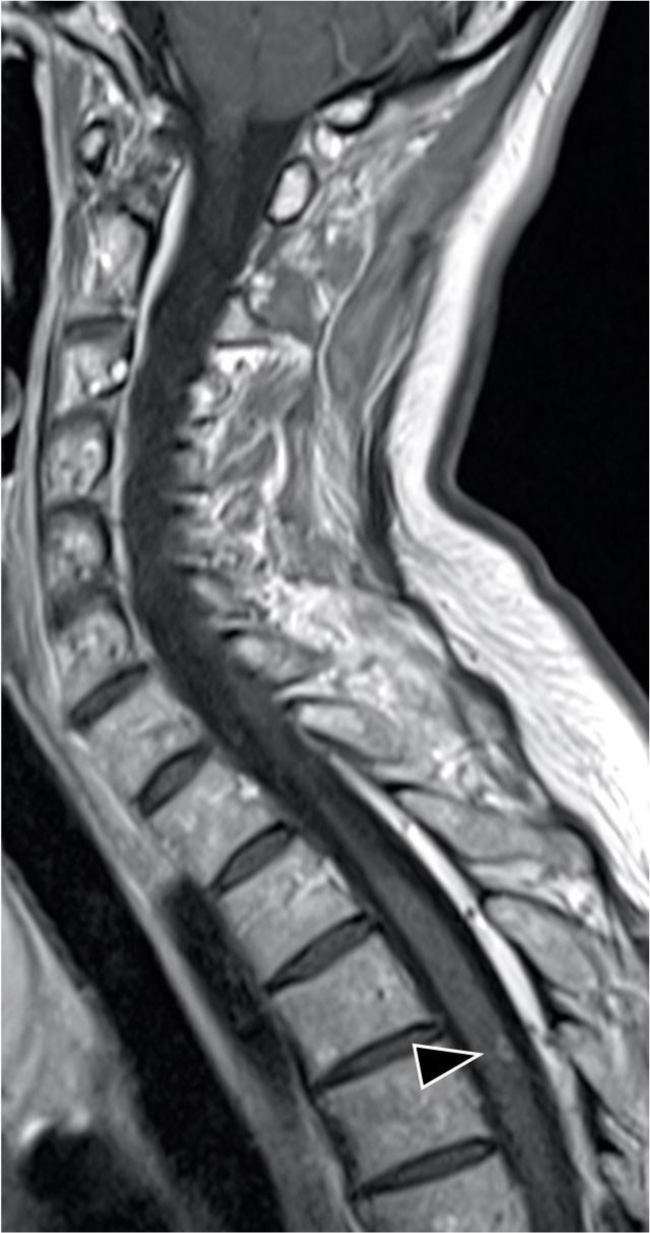
Radiology findings: Magnetic resonance (MR, 1.5 TESLA) images of the brain, spinal cord, and optic nerves showed multiple hyperintense lesions (Figs. 1 and 2), four in the brain and two in the spinal cord; Th4 and Th5 were contrast-enhanced (Fig. 3). New lesions were found in comparison to 2015, while some had decreased in size. Computed tomography (CT) of the abdomen showed hepatosplenomegaly and fibrotic changes in the renal parenchyma. Echocardiogram was found normal. The nerve conduction study showed sensory polyneuropathy in the legs. Electromyography showed no neurogenic or myogenic pathological changes.
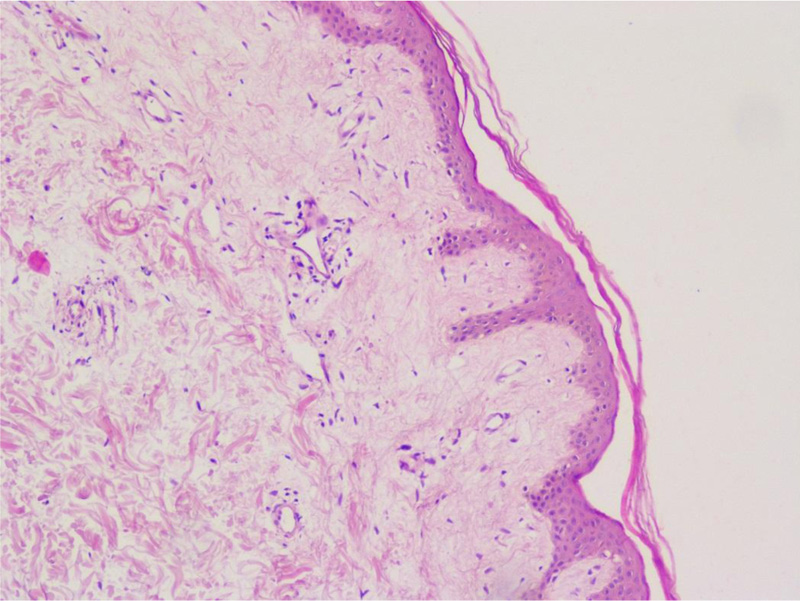
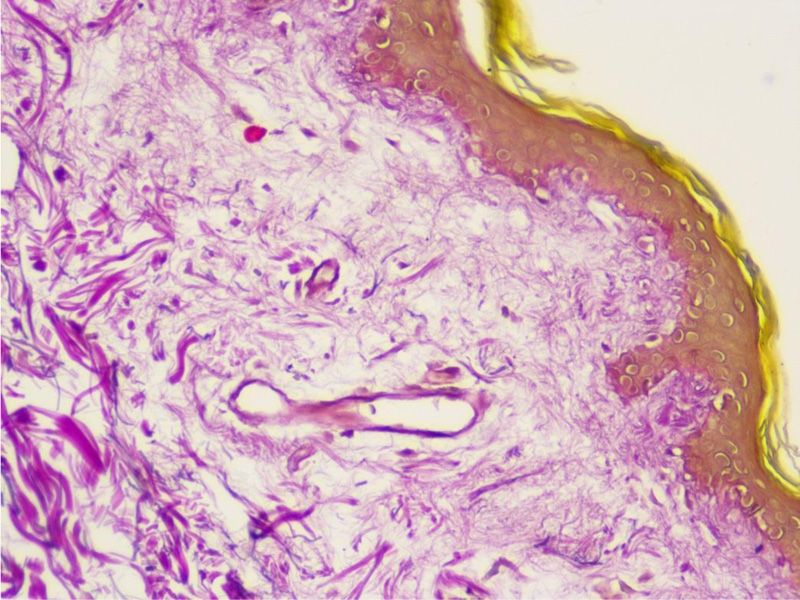
On the fifth hospital day, a skin biopsy was performed, which confirmed the diagnosis of scleroderma (Figs. 4 and 5).
The patient had received pulse therapy with methylprednisolone a few months before the hospitalization, which provided slight improvement; she felt stronger and her muscle strength improved. However, the improvement was temporary, and she started to suffer from diffuse abdominal pain. At the time of hospitalization, the patient was started on plasma exchange, but after the second plasma exchange procedure, her condition rapidly deteriorated. She developed colitis with bloody diarrhea, febrile temperature, abdominal pain, fatigue, and right-sided hydrothorax that was punctured. The patient developed pulmonary edema, followed by endotracheal intubation. The patient's overall condition stabilized, but she was unable to breathe without artificial lung ventilation via tracheostoma and could not change body position without assistance. The patient received symptomatic and antibacterial therapy, but due to her severe condition, specific immunosuppressive treatment was not initiated. The patient consulted a therapist, intensive care physician, cardiologist, infectologist, rheumatologist, surgeon, pulmonologist, gastroenterologist, and ophthalmologist. Despite intensive multidisciplinary treatment, the condition of the patient deteriorated, and one month later, she died.
3. DISCUSSION
Systemic sclerosis is a multisystem autoimmune collagen disease that manifests in the skin, lungs, heart, kidneys, gastrointestinal tract, musculoskeletal system, and peripheral, central, and autonomic nervous systems. It is characterized by small blood vessel vasculopathy and fibrosis of the skin and internal organs. Fibrosis is caused by the increased production of collagen, fibronectin, and glycosaminoglycans. Endothelial cell hyperplasia and hypersensitivity of blood vessels to vasoconstrictor stimuli contribute to vasculopathy. This form of vasculopathy mainly affects small arteries, arterioles, and capillaries, causing tissue ischemia with recurrent episodes of reperfusion [9, 11]. In limited systemic sclerosis, the face and limbs distal to the knees and elbows are affected. In diffuse systemic sclerosis, the trunk and proximal parts of the limbs are affected, as in our patient’s case [5, 11].
Systemic sclerosis is expressed in various symptoms of the internal organs, joints, and nervous system [7]. In our presented case, the disease started with changes in the skin; symptoms from the gastrointestinal, cardiovascular, and central nervous systems followed over time. Neurological involvement in systemic sclerosis is rare; it more often involves the peripheral nervous system [1]. Amaral et al., in a systematic review, described 442 cases with peripheral nervous system involvement, including peripheral sensorimotor polyneuropathy, sensory neuropathy, carpal tunnel syndrome, multiple mononeuropathy, brachial and lumbar plexopathy, cranial nerve involvement, and myopathy. In much fewer cases (177 cases), central nervous system involvement was described, including headaches, seizures, cognitive impairment, stroke, transverse myelitis, hemiparesis, spinal cord compression, and other CNS expressions [1].
As can be seen, systemic sclerosis is a heterogeneous disease with various symptoms, and it is challenging for clinicians to diagnose it early. There are criteria to help make the diagnosis earlier, designed by the EU Scleroderma Trials and Research group (EUSTAR) [4]. Diagnostic criteria for diffuse cutaneous systemic scleroderma include Raynaud's phenomenon (objective documentation) plus either nailfold capillary pattern or selective autoantibodies, or Raynaud's phenomenon (subjective only) plus both nailfold capillary pattern and selective autoantibodies as well as proximal cutaneous changes in the arms and legs proximal to the elbows and knees, chest, abdomen, and/or back [4]. Our patient had proximal cutaneous changes, Raynaud's phenomenon (subjective only), and positive antinuclear antibodies. Unfortunately, we did not perform a capillaroscopy, which is included in the diagnostic criteria, although a skin biopsy was done, which confirmed the scleroderma. Skin biopsy is not required for the diagnosis of systemic scleroderma; it is recommended in cases of diagnostic doubt of other scleroderma-like disorders and used for prognostic value and research purposes [12]. Van Praet et al. noted skin biopsy to be a useful diagnostic marker for systemic sclerosis, but it having low sensitivity [13].
As mentioned above, neurological symptoms are rare in systemic sclerosis. Nevertheless, recent studies have shown hyperintense lesions on magnetic resonance imaging of asymptomatic patients [1, 2, 4, 10, 14-16]. Mohamed et al. showed that lesions in the brain are not rare in patients with systemic sclerosis irrespective of disease duration, patient age, neurological symptoms, or disease severity. The use of magnetic resonance imaging is advised for every patient with systemic sclerosis to detect brain and spinal cord involvement earlier [9, 15]. Rania et al. also confirmed that patients with systemic sclerosis without neurological symptoms may have central nervous system involvement [16].
The pathogenesis of white matter hyperintensity has different mechanisms, which can act separately or supplement each other. The first mechanism involves non-immune reactions, with cell-mediated activity and autoantibody production, likely accounting for isolated cases of vasospasm and associated autonomic dysfunction. The second reaction is vascular, with changes in endothelial cells and basement membrane, causing non-inflammatory microangiopathy of the vasa nervorum, with a tendency for platelet aggregation, resulting in ischemic CNS injury. The third mechanism is related to collagen metabolism, the proliferation of specific fibroblast subpopulations, and invasion into tissue and fibrosis [2, 9, 15, 16]. To better understand white matter hyperintensity, more studies are needed in the future. Our patient had hyperintense lesions in the white matter in 2015, when she had her first MRI of the brain and showed neurologic symptoms of progressive weakness in the right leg and ischemic optic neuropathy in the right eye. However, the diagnosis was still unclear, and thus, no specific treatment was prescribed.
Management of systemic sclerosis includes early diagnosis of the disease, early recognition of internal organ involvement, and early identification of patients who are at risk of developing new organ complications [17]. Treatment depends on the severity of the disease, system involvement, and clinical manifestations. Treatment includes immunomodulatory and immunosuppressant therapies [7]. The updated treatment recommendations of 2016 do not mention treatment for nervous system involvement; the use of methotrexate or cyclophosphamide for systemic sclerosis skin manifestations is recommended [17]. Amaral et al., in their systematic review, reported cyclophosphamide, corticosteroids, azathioprine, and methotrexate as the most used drugs for systemic sclerosis with neurological manifestations [1]. There is conflicting data on the use of immunosuppressive therapy for the treatment of neurological symptoms in systemic sclerosis, but corticosteroids have demonstrated positive effects, as reported in Amaral et al.’s systematic review [1]. Our patient had received corticosteroid therapy and had shown improvement, but unfortunately, she did not continue tapering the oral corticosteroids.
There are publications regarding plasma exchange therapy for systemic sclerosis. Harris et al. found positive treatment effects with therapeutic plasma exchange, such as improved symptoms and laboratory markers. They described both long-term and short-term treatments. Long-term treatment was characterized as safe and well-tolerated and exhibiting improved symptoms, with the disease being stabilized. Furthermore, they did not find serious complications with plasma exchange treatment; most complications were related to the venous catheter [18]. The guidelines for plasma exchange therapy in systemic sclerosis cover mild and short-term complications [19]. Shemin et al. reported that, in some rare cases, abdominal pain was present and fever was a frequent complication during plasma exchange therapy [20]. Basic-Jukic et al. considered plasma exchange as a safe therapy [21]. Our patient, after the second plasma exchange procedure, had colitis with fever and hydrothorax, and the treatment was discontinued. We did not find any publication that mentioned colitis or hydrothorax as a complication of plasma exchange therapy, nor did we find any other reason for the patient's health to rapidly deteriorate during this therapy.
The prognosis for systemic sclerosis depends mainly on the severity of internal organ involvement, as noted by Sobolewski et al.; they observed that in the first three years of the disease, the involvement of internal organs was faster, indicating the importance of early recognition and treatment, which is critical for a good prognosis [22]. Nikpour et al. described frequent causes of death in systemic sclerosis and found that the primary cause of death was pulmonary and interstitial lung disease. One defining predictor of mortality was cardiopulmonary involvement, and their study reported 78% 10-year survival [23]. However, Pokeerbux et al. reported 71.7% 10-year survival [24]. Elhai et al. reported that in 30% of deaths of systemic sclerosis patients, the cause was heart disease, and high mortality rates were associated with respiratory failure and lung infections [25]. As mentioned above, our patient had heart failure and respiratory insufficiency, which increased the risk of mortality; furthermore, she had not received specific therapy over the years. According to Rubio-Rivas et al., among systemic sclerosis patients, 57% of deaths were lung-related, as the case with our patient [26]. We could not find any study that mentioned neurologic symptoms as a mortality risk factor, nor could we find neurological symptoms associated with a higher mortality rate. However, Poudel et al.’s study noted that patients with systemic sclerosis were two to five times more likely to die compared to the general population [27].
We should always keep in mind this heterogeneous disease if the patient has more than one system involved, especially the nervous system, skin and internal organs.
CONCLUSION
This clinical case shows the complexity and heterogeneity of systemic sclerosis and the importance of multidisciplinary teamwork. Systemic sclerosis is a challenging diagnosis for any clinician, but early detection of organ involvement and early treatment are critical for patients. But it is not always easy to detect scleroderma in the early stage of the disease and start treatment, as we saw in our case. It is important to follow up a patient and perform magnetic resonance imaging of the brain and spinal cord for each patient at the onset of the disease to detect early CNS involvement. In the future, we need more studies regarding systemic sclerosis, its treatment and associated neurological involvement.
AUTHORS’ CONTRIBUTIONS
EP, DP, LJ and GK wrote the first draft of the manuscript. All authors reviewed and edited the manuscript, made substantial contributions to the design of the work, and approved the final version of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Ethical approval for this study protocol was obtained from the regional ethics review board of the Riga East University Hospital Support Fund Medical and Biomedical Research Ethical Committee Nr.6-A/21; 05.08.2021.
HUMAN AND ANIMAL RIGHTS
No animals were used for studies that are the basis of this research. All the human procedures used were in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
Informed consent was obtained from the patient for educational use of the below-mentioned data, and no personal patient information has been disclosed.
STANDARDS OF REPORTING
CARE guidelines and methodologies were followed in this study.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available from the corresponding author, [E.P], on special request.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

