All published articles of this journal are available on ScienceDirect.

E200k Familial Creutzfeldt-Jakob Disease Presenting with Subacute Multiple Cranial Neuropathy
Authors Info & Affiliations
Abstract
Unusual clinical presentations in patients with E200K familial Creutzfeldt-Jakob Disease (fCJD) have been rarely reported. Herein, we described a case of E200K fCJD presenting with subacute cranial multiple neuropathy, initially suspected to be paraneoplastic or due to a leptomeningeal carcinomatosis, considering the neoplastic comorbidity of the patient. Surprisingly, brain MRI was highly suggestive of CJD. Brain histological examination confirmed the diagnosis. Genetic tests led to the definite diagnosis of E200K fCJD. To the best of our knowledge, the current case provides the first report of a histologically-confirmed E200K fCJD starting with cranial multiple neuropathy and may widen the spectrum of the clinical variability of CJD, also in its genetic variant. Unusual presentations may lead, as in this case, to incorrect diagnostic hypothesis and unuseful therapeutic attempts in the first phase of the diagnostic process. Also in the genetic variant of CJD, brain MRI demonstrated a very high sensitivity to detect the typical abnormalities since the earliest phases of the disease.
1. INTRODUCTION
Several unusual presentations in patients with E200K familial Creutzfeldt-Jakob Disease (fCJD) have been described in a recent review written by Cohen et al. [1]. Herein, we report the case of a histologically confirmed E200K fCJD presenting with a subacute multiple cranial neuropathy.
2. CASE REPORT
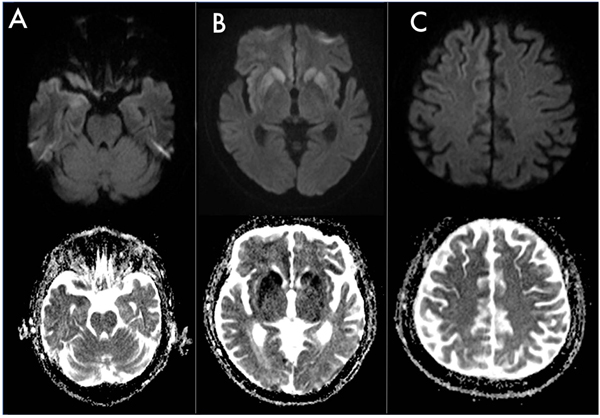
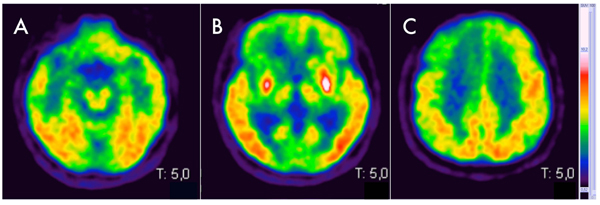
Here, we report the case of a 77-years old man with a metastatic colic cancer diagnosed two years before, who developed a multiple cranial neuropathy with a subacute onset, characterized by marked right eyelid ptosis, horizontal gaze palsy, left facial nerve palsy, hypophonia, dysarthria and dysphagia, associated to a left pyramidal-cerebellar syndrome. In particular, right eyelid ptosis and horizontal gaze palsy appeared one month before the admission in our department and had a rapid progressive course, becoming evident in one week. Two weeks later (thus, two weeks before the admission) the patient developed a left facial peripheral nerve palsy, followed by hypophonia, dysarthria and dysphagia that rapidly worsened, leading to the hospitalization. A mild left pyramidal-cerebellar syndrome characterized by limbs weakness and segmental dysmetria was detected at the neurological examination and not complained before by the patient. At the neuropsychological evaluation, Mini Mental Status Examination (MMSE) and Frontal Assessment Battery (FAB) revealed scores of 29/30 and 17/18 respectively, thus supporting preserved cognitive functions. A leptomeningeal carcinomatosis or a paraneoplastic multiple cranial neuropathy was initially suspected although total body TC/18F-fluoro-D-Glucose Positron Emission Tomography (FDG-PET) performed 3 months before the onset of neurological signs showed a steady neoplastic situation, with no increasing tumor size of both primitive cancer and pre-existing metastasis, nor detection of other sites of pathological tracer accumulation. The initial diagnostic work-up included electroencephalogram (EEG), to evaluate if the above mentioned left pyramidal syndrome, had a lateralized electroencephalographic correlate. Electroencephalogram revealed only mild and bilateral anterior slowing. Cerebrospinal Fluid (CSF) analysis included cytochemical analysis, that demonstrated the presence of 2 cell (lymphocytes)/mm3 (range 0-5 cells/mm3) with proteins and glucose levels within normal limits (30 mg/ml, normal range 15-45 mg/ml and 45 mg/dl, normal range 40-70 mg/dl respectively). Neither intrathecal synthesis of IgG nor oligoclonal bands were detected by immunochemical and immunoblot analysis respectively. Cytology showed normal mature lymphocytes with no nuclear and cytoplasmic abnormalities. Surprisingly, brain MRI showed FLAIR hyperintense signal involving right temporo-insular cortex, paramedian frontal cortex, putamen and caudate nuclei bilaterally with DWI hyperintensity seen at high b values in the same areas, with corresponding hypointensity on ADC map. The brainstem didn’t show signal abnormalities but appeared slightly atrophic (Fig. 1). Taken as a whole, MRI features were suggestive of CJD. Diagnostic work-up was completed with brain FDG-PET, that revealed cortical bilateral (but more pronounced on the right side) hypometabolism, involving temporopolar, fronto-polar, frontal dorso-ventrolateral, fronto-mesial and anterior cingulate cortices (Fig. 2). These features were associated with the evidence of bilateral putaminal hypermetabolism. Taking into account neurological features and the putaminal hypermetabolism, atypical for CJD, and the neoplastic comorbidity, a therapeutic attempt with Intravenous Immunoglobulins (IVIG, 0,4 mg/kg/die for 5 days totally) was performed, with no clinical improvement. Onconeural intracellular (anti-Hu, anti-Ri, anti-Yo, anti-Ma) and surface antigens (anti-NMDA, VGCK, GABA-a, GABA-b) antibodies were tested on CSF, with negative result. In the following month, the patient rapidly worsened and developed a severe cognitive impairment, finally evolving into mutism. Prominent oro-buccal dyskinesia also characterized this phase of the disease. Serial electroencephalograms were performed, with the final evidence of typical periodic waves. In keeping with the notion of massive neurodegeneration [2], high tau (2269 pg/ml, normal range <400 pg/ml; ELISA, INNOTEST KIT, Fujirebio, Europe, Gent, Belgium) and mildly increased hyperphosphorylated-tau protein (69 pg/ml, normal range <40 pg/ml, ELISA, INNOTEST KIT, Fujirebio, Europe, Gent, Belgium) levels were detected in CSF, that turned positive also for 14-3-3 protein detection. The patient died one month later. Brain histological examination confirmed the diagnosis of CJD and PRNP gene sequencing revealed a Methionine-Methionine (MM) polymorphism on codon 129 and the E200K mutation, leading to the diagnosis of E200K fCJD. No evidence of metastatic involvement of cranial nerves and meninges was found at the histologic examination. Since, the patient was adopted after birth, data concerning his family history are not available.
3. DISCUSSION
The wide phenotypic variability of fCJD clinical onset has been previously reported by Chapman et al. [3] and Cohen et al. [1]. The risk of misdiagnosis and useless treatments comes down to unusual atypical presentations, particularly during the earliest stages of the disease. To the best of our knowledge, the current case provides the first report of a histologically-confirmed E200K fCJD starting with multiple cranial neuropathy. In our case, the neoplastic comorbidity and the initial lack of cognitive issues were misleading, while the typical cortical, putaminal and caudate restricted diffusion prompted us to investigate and identify this genetic variant of CJD. As previously reported, atrophic changes in the cerebellum and brainstem may cause elevated diffusivity leading to low, rather than high, signal in Diffusion Weighted Images (DWI) sequences and high Apparent Diffusion Coefficient (ADC) values. Moreover, DWI scans, set specifically to detect diffusion restriction may miss brainstem and posterior fossa changes [4]. Further, the absence of signal abnormalities in the brainstem on T2-FLAIR and T1SE sequences may be related to the low spatial resolution of these MRI sequences, not able to detect the pathological involvement of cranial nerves and/or brainstem nuclei.
The involvement of peripheral nervous system in human prion disease has not been widely investigated. In one paper [5], electrophysiologic examinations confirmed peripheral nervous system damage in patients with sporadic CJD and revealed also preclinical peripheral nervous system impairment. Furthermore, in another report [1] neuronal loss, gliosis and Prionic Protein (PrP) deposits were observed in the vestibular and cochlear nuclei of a patients presenting with sesnorineural hearing loss. Similarly, cranial nerves involvement in our patient could be explained by the direct deposition of PrP in oculomotor, VII, IX and X brainstem cranial nerves nuclei. Unfortunately, no specific histo pathological examinations for Prp deposition in the cranial nerves and nuclei has been performed in our patient. Never theless, the absence of hystologic abnormalities suggestive of metastatic involvement of cranial nerves and meninges allows us to formally exclude a metastatic spreading in the brainstem as cause of the cranial multiple neuropathy.
18F-FDG-PET have been studied in prion disease although there are few studies with autopsy-confirmed cases and currently it is not routinely used in the diagnostic evaluation. Cortical hypometabolism in human prion diseases represents a well-known phenomenon, neuropathologically related to neuronal loss, astrocytosis and/or spongiosis. Conversely, hypermetabolism in 18F-FDG-PET seems to be related with microglial activation and neuroinflammation, as demonstrated in sporadic CJD [6]. This pattern has been recently described in limbic and mesolimbic structures [7] but no cases of putaminal hypermetabolism, as shown in our patient, have been currently reported.
CONCLUSION
This case widens the broad phenotypic variability of fCJD, potentially leading to misdiagnosis and unuseful therapeutic attempts. Furthermore, it confirmed MRI highest sensibility and specificity for an early diagnosis, also when clinical features at the onset are atypical for prion disease.
LIST OF ABBREVIATIONS
| fCJD | = familial Creutzfeldt Jacob disease; |
| CSF | = Cerebrospinal Fluid; |
| 18F-FDG-PET | = 18F-fluoro-D-Glucose Positron Emission Tomography; |
| DWI | = Diffusion Weighted Images; |
| ADC | = Apparent Diffusion Coefficient; |
| MM | = Methionine-Methionine. |
AUTHORS CONTRIBUTION
All authors (Lapucci C., Romano N., Boffa G., Saitta L., Nobili F., Mancardi G.L., Mandich P., Grandis M.) made a substantial contribution to the design, data collection and analysis of the research and the drafting of the manuscript and reviewed and accepted the contents of the manuscript prior to its submission.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Informed consent for publication was obtained by a next-of-kin of the patient after his death.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

