All published articles of this journal are available on ScienceDirect.

Are Temporal Differences in GDNF and NOS Isoform Induction Contributors to Neurodegeneration? A Fluorescence Microscopy-Based Study
Authors Info & Affiliations
Abstract
Background:
Specific factors in Parkinson’s disease have become targets as to their protective and degenerative effects. We have demonstrated that cytokines and PD-CSF detrimentally affect microglia and astrocyte growth. While glial cell-derived neurotrophic factor (GDNF) has been recognized as a possible neuron-rescue agent, nitric oxide synthase (NOS) has been implicated in neurodegenerative processes.
Objective:
To demonstrate that glial cell activation, cytokine production, and NOS induction, play an intimate role in the loss of dopaminergic signaling, via mechanisms that are a result of inflammation and inflammatory stimuli.
Methods:
Study animals were sacrificed following endotoxin treatment and tissue sections were harvested and probed for GDNF and NOS isomers by fluorescence deconvolution microscopy. Fluorescence was mapped and quantified for each probe
Results:
An immune cell influx into ‘vulnerable’ areas of the brain was seen, and three NOS isomers, inducible (iNOS), neuronal (nNOS) and endothelial (eNOS), were synthesized in the brains, a finding which suggests that each isomer has a role in neurodegeneration. eNOS was found associated with blood vessels, while iNOS was associated with glial and matrix cells and nNOS was located with both glia and neurons. Following endotoxin treatment, serum levels of nitric oxide were higher at 6-8 hours, while tissue levels of NOS were elevated for much longer. Thus, induction of NOS occurred earlier than the induction of GDNF.
Conclusion:
Our findings suggest that the protective abilities of GDNF to combat neural destruction are not available rapidly enough, and do not remain at sufficiently high levels long enough to assert its protective effects. (250).
INTRODUCTION
Li and colleagues (1995) reported that glial cell line-derived neurotrophic factor (GDNF) had the ability to “rescue motorneurons” from cell death and suggested that it had a role in amyotrophic lateral sclerosis [1]. Further evidence for the protective roles of GDNF were described in a study investigating cerebral cortex ischemia and spinal cord injury-associated inflammation [2, 3]. Subsequently, GDNF and other neurotrophic factors, received much attention, with a major focus on the therapeutic possibilities in Parkinson’s disease (PD) of these and similar compounds [4] especially with the underlying problems involving oxidative stress [5, 6]. Following reports of an intimate role of oxidative stress in Parkinson’s disease, pathologic mechanisms of mitochondrial dysfunction and autophagy associated with LPS treatment and ensuing cell death were published [7-9]. These strong links of inflammation being involved in the onset of PD, including neuroinflammation associated glial activation and progression to severe neurodegeneration and similar diseases, have since been frequently reported [10, 11].
Research has been both basic and clinical, with a focus to ‘discover’ anti-inflammatory treatments for PD, as it has been postulated that non-steroidal anti-inflammatory drugs (NSAIDS) might have some benefit, with positive outcomes having been reported in animal models [12, 13]. However, there have been conflicting results as to the benefits of NSAIDS, mainly due to the complexities of neurodegenerative illnesses [14].
A recent study investigating the protective effects of GDNF, reported that an intravenous infusion of gene-modified cord blood CD34+ cells afforded some protection against cerebral ischemia [15], while adverse effects of neurotrophin therapy in patients has also been reported so evidence supporting the benefits of neuro-proteins and anti-inflammatories against intra cranial cell death is inconclusive [4].
The underlying and inter-dependent pathways and mechanisms that combine to drive neurons to non-retrievable and inevitable death have been fairly well established by Naoi et al, [16]. In our previously published research we had focused on identifying the interplay of these pathways following treatments with various inflammatory cytokines [17, 18]. The results suggested i) that cytokines do alter expression and aggregation of PD-associated proteins in cultured cells; ii) protein aggregations, regulations and syntheses in lipopolysaccharide endotoxin (LPS) treated rat brains are both time dependent and area and cell specific, thus mimicking our findings in cultured cells; iii) resilience to ‘toxic’ assault is different among cell types in terms of time dependency and culpability; and iv) expression and regulation of protective and regulatory markers like GDNF and NOS, does not follow a time line that is beneficial to the preservation of neuron function and viability.
iNOS being present in neurodegeneration has been linked to detrimental outcomes in neurons, with the theory being that a selective iNOS inhibitor or NSAID, might be a useful therapeutic intervention and be able to avert losing dopaminergic neurons [19, 20].
Our current research suggests that if time dependent factors can be manipulated so that they are present in sufficient concentrations at such a time that coincides with toxic/damaging events, or if they are available over an extended period of time, then preservation of cognitive ability might be achievable. Such ideas have been formulated previously in PD [21], with basal ganglia and nociception changes due to reports of pain and abnormalities in nociception and mechanical thresholds having been reported [22].
MATERIALS AND METHODS
i. Sectioning and Staining of Tissues
Adult rat brains (300g Sprague Dawley; males), from untreated control, saline-injected animals and LPS treated animals (35 µg/kg), were removed, fixed, sectioned and probed with antibodies. Tagged secondary antibodies were added for visualization using deconvolution fluorescence microscopy. Images were acquired in a stepwise section thickness (0.1-0.25 µm) from the olfactory bulb and cortex following the guidelines of Paxinos and Watson [23]. The protocol for this work was approved by this institutions animal welfare committee (AWC-14-0148 Title: Long-Term Effects of LPS in Rats: A Model of Neuro-Degenerative Disease).
ii. Imaging
Sections were mounted on 25 mm glass cover slips and fluorescently probed for the three NOS isoforms. Tissue was rinsed with cold PBS and fixed with 3.7% formaldehyde (1008B, Tousimis Research Corp., Rockville, MD). Fixation was halted by 3 rinses with PBS at room temperature and blocked with 10% goat serum, then incubated for 1hr at 37oC with antibodies (1:100 to 1:1000 dilutions), incubated for 30 min. at 37oC and rinsed 2 to 3 times with 0.05% Tween 20 in PBS (TPBS). Finally, nuclei and F-actin were stained with Phallicidin (Invitrogen, Grand Island, NY, USA) diluted 1:100 in 4', 6-diamidino-2-phenylindole (DAPI; A-T rich region binding in DNA) solution. Specimens were mounted on slides with Elvanol (DuPont, Wilmington, DE). Fluorescence intensity measurements were made using pixel counts as previously described [17, 18, 24] and 3D reconstructions and rotations were performed [25].
iii. Deconvolution Fluorescence Microscopy
Deconvolution fluorescence microscopy and 3D image reconstructions of NOS isoforms, actin and nuclei were generated as previously described [17, 18]. Images were saved as TIFF files and comparative statistical analyses (one way ANOVA) of both cell count and fluorescence densities (pixel numbers) were made using Corel (Ottawa, Ontario, Canada) and SigmaStat software (SPSS, Chicago, IL, USA).
iv. NOS, Isoforms and Western Blot
To ensure that a robust inflammatory response had occurred, frozen tissues were homogenized at a ratio of 1g per 3-6 ml of buffer, first with pestle and mortar and then with an homogenizer in an ice + salt cold environment. Concentrations of iNOS, nNOS and nitrotyrosine were measured using specific antibodies. Frozen tissue homogenates were thawed on ice and mixed with equal volumes of sample buffer (200 mM Tris-HCl (pH 6.8), 8% sodium dodecyl sulfate, 30% glycerol + 1% β-mercaptoethanol, 0.5% bromophenol blue), then heated at 95°C for 5 to 10 min. Samples were centrifuged and loaded on 8% Tris/glycine/sodium dodecyl sulfate-polyacrylamide gels for fractionation, with predetermined molecular weight standards (Novex Inc., Encinitas, CA). Protein was blotted onto nitrocellulose membranes at 4°C at 30 V and 380 mA for 150 min. After transfer, the membranes were incubated with 5% skim milk in Tris-buffered saline containing 0.05% Tween 20 for 2 h at room temperature as previously described [26].
v. Nitrotyrosine Immunoblot
Samples for immunoblot analysis were boiled for 3 min and electrophoresed on 12.5% acrylamide gels. The proteins were electrophoretically transferred to nitrocellulose sheets using a semidry apparatus. The sheets were reacted with a monoclonal mouse anti-nitrotyrosine antibody (Zymed Laboratories, South San Francisco, CA) and immune complexes were detected using peroxidase-labeled goat anti-mouse immunoglobulin G (KPL, Inc., Gaithersburg, MD; 20. Fig. (1) as a marker, to show that oxidative stress had resulted due to LPS treatment [26].
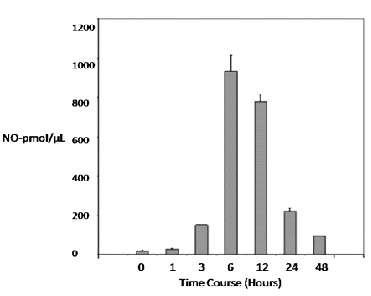
RESULTS
Our data show that the circulating level of nitric oxide (NO) following a single 35µg/kg dose of LPS peaked at 6-12 hours and remained higher than basal levels up to 48 hours after endotoxin treatment. This reveals that there is a sustained inflammatory response and that the inflammatory event is achieved with a considerable intensity (Fig. 2).
The graph on the left (Panel A - Cell Types) shows that more lymphocytes are present up to and beyond 6 hours, with the cytokine concentration also increasing dramatically (Panel A - Proteins), particulary TNF α and interleukin-6, which both have been implicated in neurodegeneration. Although the GDNF concentration also increases, the amount is somewhat modest, possibly not sufficient to overcome destructive actions of cytokines.
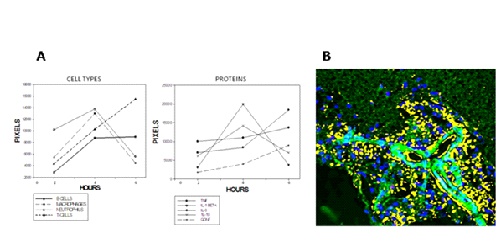
Panel B Fig. (2) is a fluorescence image of cytotoxic t-cells (yellow), 12 hours after LPS administration and reveals a substantial number clustered around an arteriole, whuch suggests the involvement of the blood-brain barrier in inflammation, and possibly an influx of circulating factors and cells into the brain.
As shown in Fig. (3), LPS treament resulted in a substantial, sustained increase in inducible nitric oxide synthase (iNOS) over 12 hours (2 and 8 hours shown), with iNOS clearly concentrated along nuclei (blue) of sensory cell tracts.
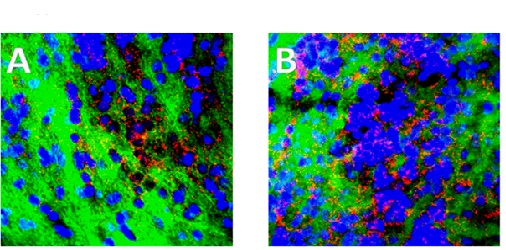
In our studies we used the cortex as a “control”, but concentrated on the olfactory bulb as a loss of olfaction is an early indicator of PD. However when comparing the cortex with the olfactory bulb, both areas showed a substantial increase in iNOS, attesting to a major and prolonged (>48 hours) inflammatory response to LPS administration (cortex images not shown).
Earlier work [27] focused on cytokines and PD-associated proteins (tau, α-synuclein and ubiquitin) and we reported a major role for microglia and astrocytes in neurodegeneration, as well as the likely involvement of a compromised blood brain barrier (BBB). The BBB has been mentioned frequently in the literature as a culprit in neurodegenerative disease, so we looked at endothelial nitric oxide synthase. The images in Fig. (4) show eNOS around vessels, 0, 2 and 8 hours after LPS treatment, with a multitude of capillaries clustered around an artery (Panel A), an arteriole (blue arrow) and a venule (yellow arrow) in panel B, and two arterioles in panel C. The images reveal a ‘separation’ of the eNOS from a peri-medial location to an adventitia area 2 hours after treatment, but no such structural change is seen at time zero, or after 8 hours.

Therefore, we modeled our images to have a better view of eNOS (red) association with vessels, and constructed rotatable, 3D models, here showing (Fig. 5) eNOS around a venule (panel A) and an arteriole (panel B) in a section of olfactory bulb, demonstrating the specificity of this isoform to a particular cell type. Smaller vessels can also be seen coursing throughout the tissue. The connective tissue matrix has been removed for clarity. Thus, we have demonstrated very specific NOS isoform associations.

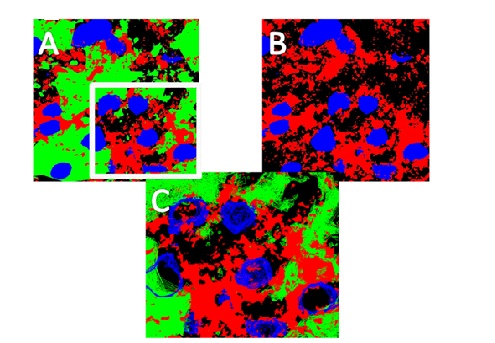
Fig. (6) is a compilation of images to show different image renditions that give us the ability to see juxta positiong of cells and markers. Panels A, B, C show combinations of nNOS (red), connective tissue (green) and nuclei (blue) as a complete-field stack of all three probes (A), then with the connective tissue (green) removed (B), and finally with part of the image ‘wire-framed’ at a higher magnification to allow us to interprete juxta-positioning of nuclei and nNOS to assess specificity.
DISCUSSION
In 2006 Arai and colleagues [28] stated, “The cause of nigral dopaminergic neuronal cell death in PD and its underlying mechanisms remain elusive” and our studies have shown a number of interesting findings with microglia cultures, which can synthesize PD-associated proteins in response to exposure to various cytokines and CSF from PD patients and cultured astrocytes, which are also susceptible to cell death and PD-associated syntheses following similar treatments. These reults lead us to believe that astrocytes are also intimately involved in the neurodegenerative process [17].
Work identifying cytokines and immune cells in an animal model pointed to the likelihood that the blood brain barrier was involved and possibly compromised in the inflammatory process leading to neurodegneration [27]. Circulating cytokines and immune cells have been reported as major contributors in the inflammatory episode, or episodes, that lead to the start of the neurodegeneration cascade, a mechanism that has been accepted for some time by neurologists and neuroscientists [24, 29, 30]. We had hypothesized that either one massive inflammatory episode, or a number of additive, smaller events [18], could result in continuing and ultimately disastrous neuronal loss, an idea that has now been supported by recent studies [31, 32] and was also shown by Qin et al. [33].
To uncover pro-degenerative factors in this complex, multi-faceted neurodegeneration cascade, we determined levels and locations of NOS isoforms, as these mediators have been implicated in neurodegeneration and are thought to play an important role in the progression of neuronal loss, while the protective peptide GDNF is overwhelmed by continuing inflammation and associated syntheses of pro-inflammatory cytokines, influxes of peripheral immune cells and activiation of microglia [34-36].
Our results support the involvement of NOS, because blood vessel-associated eNOS is ‘disrupted’ 2 hours after LPS treatment, becoming less associated with the intima and media of arterioles and venules, but resuming a normal distribution pattern after a few more hours. This change might indicate an initial ability to overcome an inflammatory assault, even though high levels of TNFα and the presence of cytotoxic t-cells, and only low levels of protective GDNF, are present.
A recent paper [36] stated, “We seem to be missing some key parts of the jigsaw, the trigger event starting many years earlier in the disease process, and what we are looking at now is merely part of a downstream process that is the end stage of neuronal death”. We published similar views (Presented at Experimental Biology 2013, Boston), that of an early event or events leading to continuing neuro-defense problems that are eventually overwhelmed and multiple signaling attributes, such as neuro-muscular and olfaction, are lost [24].
In their paper, Dexter and Jenner [37] stated that all these factors were only “parts of the jigsaw” and that “key parts” were missing. If we are unable to construct a complete picture of the early problems associated with PD that can be targeted by treatment, then the inevitable point of no return is reached. It behooves us therefore to search for targets in this complex cascade in order to at least slow disease progression and, although there is hope with anti-inflammatory treatments [38], nitric oxide attenuation of inflammation [39] and GDNF [40], success so far has been limited. New studies should probably be directed at a multi-targeted approach to include reducing inflammation, reducing cytokine levels and deactivating glial cells, while increasing endogenous levels of GDNF and attenuating NOS in order to protect the blood-brain barrier [41, 42]. Studies have been performed with lengthy treatments [43] as short treatments do not result in a cessation of neurological damage and disease progression [44], but acute therapies should not be abandoned.
Recently a paper was published stating that microglia, astrocytes, T-cells and barrier disruption all play a part in neurodegeneration, and that the “mechanism of neuroinflammation regulation is not fully deciphered” [45]. Once an understanding of the many problems associated with this complex neurodegenerative cascade has been elucidated, it should become easier to at least slow PD progression, while hoping neuronal signaling mechanisms can be restored, thus giving respite to PD sufferers as well as to their caregivers and families.
CONCLUSION
There is compelling evidence that neurodegeneration in Parkinson’s disease has multiple factors, mechanisms and pathways all combining to bring about dementia, loss of olfaction and gait problems. There is also data which strongly suggest that the many factors are both peripheral and central, and include circulating cytokines, immune cells, microglia and astrocyte activation, mitochondrial dysfunction, protein syntheses, free radicals and protective factors. This work has noted that astrocytes are probably more involved than previously thought; that blood vessel structure-function relationships change; that GDNF is not made fast enough or in enough quantities to continue to overcome degenerative assault; and that there is a great deal of specificity as to cytokines, nitric oxide, cell types and brain area.
LIST OF ABBREVIATIONS
| GDNF-Glial | = Cell-line derived neurotrophic factor |
| LPS | = Lipopolysaccharide endotoxin |
| NOS | = Nitric oxide synthase |
| PBS | = Phosphate buffered saline |
| PD-CSF | = Parkinson’s disease patient cerebrospinal fluid |
| PD | = Parkinson’s disease |
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

