All published articles of this journal are available on ScienceDirect.

Potential Role of Caffeine in the Treatment of Parkinson’s Disease
Abstract
Parkinson’s disease [PD] is the second most common neurodegenerative disorder after Alzheimer’s disease, affecting 1% of the population over the age of 55. The underlying neuropathology seen in PD is characterised by progressive loss of dopaminergic neurons in the substantia nigra pars compacta with the presence of Lewy bodies. The Lewy bodies are composed of aggregates of α-synuclein. The motor manifestations of PD include a resting tremor, bradykinesia, and muscle rigidity. Currently there is no cure for PD and motor symptoms are treated with a number of drugs including levodopa [L-dopa]. These drugs do not delay progression of the disease and often provide only temporary relief. Their use is often accompanied by severe adverse effects. Emerging evidence from both in vivo and in vitro studies suggests that caffeine may reduce parkinsonian motor symptoms by antagonising the adenosine A2A receptor, which is predominately expressed in the basal ganglia. It is hypothesised that caffeine may increase the excitatory activity in local areas by inhibiting the astrocytic inflammatory processes but evidence remains inconclusive. In addition, the co-administration of caffeine with currently available PD drugs helps to reduce drug tolerance, suggesting that caffeine may be used as an adjuvant in treating PD. In conclusion, caffeine may have a wide range of therapeutic effects which are yet to be explored, and therefore warrants further investigation in randomized clinical trials.
1. INTRODUCTION
Parkinson’s disease [PD] was named after James Parkinson, an English physician who described it as Shaking Palsy in 1817 [1]. It is the second most common neurodegenerative disorder after Alzheimer’s Disease [AD], affecting 1% of population over the age of 55 [2, 3]. Clinical manifestations of PD includes a triad of motor symptoms (bradykinesia, rigidity, and resting tremor) and a range of neuropsychiatric and autonomic manifestations [4]. The precise aetiology of PD is unknown but studies have shown it to be multifactorial with an underlying genetic and environmental component. A number of single gene defects have been identified and these account for only 10% of PD cases; with sporadic forms accounting for the remaining cases [5]. The underlying neuropathology seen in PD is characterised by progressive loss of dopaminergic neurons in the substantia nigra pars compacta [SNc] with the presence of Lewy bodies [LBs] and Lewy neurites [LNs] which are primarily composed of aggregates of alpha-synuclein [α-synuclein] [6].
Emerging data from epidemiological studies suggests that PD is less common in caffeine users compared to the general population [7, 8]. Furthermore, both in vitro and animal studies indicate that caffeine in low to moderate doses has neuroprotective effects [9]. Data suggests that caffeine antagonises adenosine A2A receptors as well as prevents aggregation of α-synuclein [10]. This literature review aims to explore the neurophysiological effects of caffeine and its potential role as an adjuvant in the pharmacotherapy of PD.
1.1. Risk Factors
Although the aetiology of PD remains unknown, there are a number of important risk factors that play an important role in the development of PD.
1.1.1. Age and Gender
PD mostly affects individuals over the age of 50, although rare exceptions in younger individuals are reported. The risk of developing PD increases with age due to changes in neuronal circuitry and an increase in inflammatory response in neurons. Age-related cognitive decline and olfactory dysfunction are changes observed in approximately 80% of individuals in the early stages of the disease [11-13]. Such changes are associated with decreased neuronal density and cholinergic and serotonergic neurotransmitter concentrations in areas such as the locus coeruleus which may attribute to loss of olfaction [14]. In addition, PD is more prevalent in males due to influences of genetic, environmental, and physiological factors. For instance, since the farming industry is male-dominated, a higher proportion of male farmers are likely to be exposed to high levels of pesticides, which may trigger PD-inducing neurotoxicity.
Genomic imprinting inactivates one of the two X-chromosomes in females through methylation. This process may silence a number of genes involved in the pathogenesis of PD on the X-chromosome, halving the effect of any risk alleles and thus reducing susceptibility to PD [15, 16]. In addition, higher concentrations of endogenous oestrogen in females may have neuroprotective effects since endogenous oestrogen prevents neurotoxin build-up and loss of dopam-inergic neurons in the SNC. This effect is not observed in males since oestrogen levels in healthy males are relatively lower than in females [17]. Nevertheless, the protective effect of oestrogen diminishes in postmenopausal women, and their risk of developing PD is then equivalent to males [18].
1.1.2. Family History
Monogenic forms account for less than 5% of cases of PD. Genomic variants in at least seven genes are associated with both autosomal dominant and autosomal recessive forms of PD. These genes (SNCA, LRRK2, EIF4G1, VPS35, parkin, PARK2, PINK1 and DJ1/PARK7) are reviewed extensively elsewhere [5, 19].
For instance, α-synuclein (SNCA) is central to both familial and sporadic PD. A number of missense mutations in SNCA (A30P, E46K, and A53T) increase the tendency of α-synuclein protein to form β-sheets which aggregate and form LBs [20]. Multiple copies of this mutated gene have been reported in a number of familial cases of PD [21].
1.1.3. Occupational Risks and Head Trauma
Farmers are more likely to be exposed to pesticides, which are linked to neurodegenerative disorders. A number of pesticides contain neurotoxins such as dipyridylium herbicides that are structurally similar to 1, methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine [MPTP], a fast acting neurotoxin. Such herbicides can induce parkinsonian symptoms and predispose individuals to the disease by generating reactive oxygen species which destroy dopaminergic neurons in the SNc [22, 23]. Moreover, some pesticides are known to inhibit neuronal aldehyde dehydrogenase [ALDH] activity which increases individual susceptibility to PD, particularly in individuals carrying genetic polymorphisms in ALDH2 [24].
Head injuries can cause cerebral haemorrhages. Chronic head injuries involving concussion are reported to predispose individuals to neurodegenerative disorders, including PD [25, 26]. Concussion often leads to local inflammation where pro-inflammatory cytokines cause neuronal cell death and remodelling of the brain parenchyma, which is often witnessed in autopsies of Parkinson diseased brains [27]. Moreover, brain injury triggers astrocyte activation which leads to neuronal injury and scarring since astrocytes are no longer able to maintain the integrity of the blood-brain barrier [BBB], thereby allowing toxic molecules to reach the CNS [28].
1.1.4. Lower Levels of Folic Acid
Low levels of folic acid are commonly associated with birth defects. Emerging research on rodents suggests that low levels of folic acid in diet may increase the risk of developing PD. Evidence shows that folic acid normally reduces mitochondrial dysfunction and promotes the generation of new mitochondria that may help protect neuronal cell death. Folate deficiency may lead to greater susceptibility to oxidative damage in mitochondria and apoptosis of dopaminergic neurons in PD [29]. However, at least one prospective clinical trial failed to identify any link between high folic acid intake and a reduced risk of development of PD [30].
1.2. Neurophysiology of Basal Ganglia
The basal ganglia [BG] are comprised of a number of nuclei in the subcortical region. They play important roles in the initiation of muscular movements, learning, cognition, and help to coordinate changes in posture. They are comp-rised of a number of structures, which includes the substantia nigra - a midbrain structure consisting of the pars compacta [SNc] and pars reticulate [SNr]. The SNc plays an important role within the basal ganglia in synthesising and secreting dopamine which binds to Dopamine-1 receptors [D1R] in the BG [31]. This subsequently activates GABAergic neurons favouring direct pathways, thus facilitating movement [32]. When dopamine binds to Dopamine-2 receptors [D2R], indirect pathways are activated which subsequently decreases thalamo-cortical firing and inhibits movement as illustrated in Fig (1). The striatum also projects into a number of areas which includes the GPi (contains GABAergic output neurons), the thalamic nucleus, reticular formation (affects muscle tone), and superior colliculus (affects eye movements). Alteration in these systems can be observed in PD patients, along with the bradykinesia which is a result of depletion of dopaminergic neurons in these pathways [32].
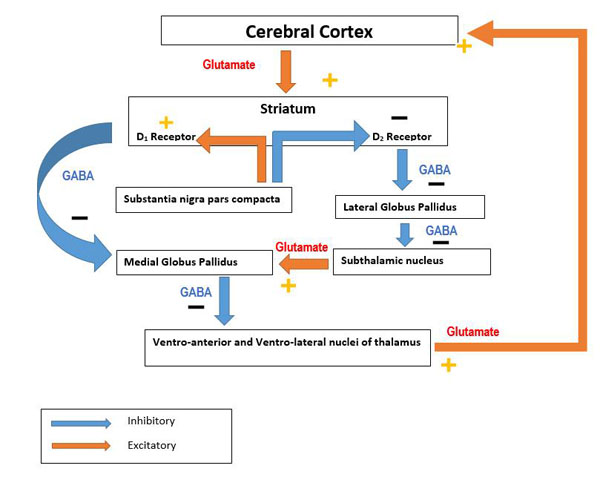
Direct and indirect pathways of Basal Ganglia in initiating motor activity. In the direct pathway, cerebral cortical input to the striatum causes activation of inhibitory neurons in the striatum which then causes an increased inhibitory output to the globus pallidus internal [GPi]. There is a decreased inhibitory output from GPi to ventral anterior [VA] and ventral lateral [VL] nuclei of the thalamus which then projects via excitatory pathways into the premotor cortex. The direct pathway is involved in regulating tonic excitation in the premotor cortex which is an area involved in planning and initiating movement. The indirect pathway is inhibitory to movement when excitatory projection from cerebral cortex facilitates inhibitory projection neurons in globus pallidus external [GPe]. These then inhibit tonic inhibitory output neurons which decreases tonic inhibition of subthalamic nucleus [STN] resulting in increased excitatory output to GPi. Excitatory input to GPi increases inhibitory output from GPi to thalamus which then decreases excitatory feedback to cerebral cortex leading to inhibition of motor activity. Dopamine promotes action of direct pathway while suppressing the activity of indirect pathway.
1.3. Pathophysiology of PD
PD occurs when the activity of neurons in GPe is reduced. This results in an increased discharge of STN neurons. The degeneration of dopaminergic SNC neurons leads to increased activity of the indirect pathway which causes an oscillation in electrical discharge to neighbouring neurons affecting the overall activity throughout the BG [34]. Pathological changes in basal ganglia results in the development of a resting tremor, loss of coordination, bradykinesia and postural changes. Rigidity is a product of dopaminergic neuron destruction in SNc which leads the striatum to over stimulate the corticospinal motor system [33, 34].
1.3.1. Molecular Histopathology of PD
The accumulation of LBs and LNs in neurons is a characteristic histological finding in PD. LBs and LNs are aggregates of α-synuclein, parkin, and synphilin-1 proteins. α-synuclein is found predominantly in the brain and expressed in mitochondria of the striatal and thalamic neurons [35, 36]. The precise role of α-synuclein is not known but a number of studies suggest that α-synuclein helps to regulate dopamine release and supply synaptic vesicles to presynaptic terminals [37]. A number if missense mutations in the α-synuclein gene (A53T, A30P, and E46K) allow the α-synuclein protein to form aggregates of β-sheets which then form toxic oligomers commonly observed in hereditary forms of PD [38, 39]. Mutated forms of α-synuclein may have prion protein-like characteristics since it can induce β-confirmation in normal α-synuclein proteins. It does so by acting as a template to promote misfolding and aggregation of normal α-synuclein which can then spread on to other regions of the brain [40]. In addition, LBs and LNs have also been reported in striatal and nigral neurons of individuals who have undergone fetal-nigral mesencephalic cell transplantation [FNMCT], further supporting the hypothesis of prion protein-like characteristic of mutated α-synuclein [40]. Long-term accumulation of mutant α-synuclein protein also leads to dementia and behavioural as well as cognitive impairment. These are characteristic features of late stages in PD [41]. In addition to α-synuclein protein, defects in the parkin gene which produces mutated ubiquitin ligase prevents removal of α-synuclein aggregates, and allows LBs and LNs to form [42]. Recently, studies reported observing ferric deposition in parkinsonian brains. It was postulated that high level of iron present in SNc may induced oxidative stress which then leads to loss of dopaminergic neurons [43].
2. NEUROPROTECTIVE AGENTS
Neuroprotection refers to the preservation of neuronal structure and function. Neuronal integrity can be compromised in diseases such as PD, and the aim of neuroprotective agents is to protect or slow down the progression of disease by delaying the process of neuronal loss. There are a number of candidates which help to reduce neurotoxicity by inhibiting neurotoxins, ROS, preserving mitochondrial function and preventing protein aggregations [44]. Neuroprotective agents include the following:
2.1. Polyphenols
Polyphenols are naturally occurring compounds found in plants that are responsible for protecting them against UVB radiation. Polyphenols have antioxidant and anti-carcinogenic effects which are yet to be explored and could potentially be used to treat a number of degenerative diseases including PD. In addition, polyphenols found in black tea are reported to inhibit α-synuclein aggregation and degeneration of dopaminergic neurons through an unknown mechanism [45].
2.2. Nicotine
Nicotine is a constituent of cigarettes and is a known potent parasympathomimetic alkaloid that acts on nicotinic acetylcholine receptors [nAchR] found in brain. Nicotine can protect nigrostriatal damage and help to improve cognitive functions according to a number of randomized controlled trials [46]. The exact mechanism of action of nicotine in nigtrostiatal pathways remains unknown but some studies suggest that nicotine binds to nAchR and activates intracellular transduction, altering calcium signalling which may help reduce neuronal injuries [46]. In addition, nicotine has been found to reduce L-dopa-induced dyskinesia [LID] in a number of animal studies but not in humans [47].
2.3. Curcumin
Curcumin is an active ingredient of turmeric. It is non-toxic and able to cross the BBB. Curcumin is capable of binding to mutant α-synuclein, thereby preventing β-aggregation [48, 49]. In doing so curcumin can help reduce LBs formation and reduce risk of developing PD. Its precise mechanism of action is not known but some suggest that curcumin acts as an amyloid breaker with anti-inflammatory effects in which it is able to reduce generation of ROS [50].
2.4. Caffeine
Caffeine 3,7-trimethylpurine-2,6-dione is a psychoactive methylxanthine extracted mainly from plant sources. It is found in a number of beverages such as coffee and tea [51]. 87% of caffeine is consumed in coffee, soft drinks and tea with an average consumption of 193mg of caffeine per day 1.2mg caffeine per kg of body per day [52]. Men and women aged 35-64 were reported to be the highest caffeine consumers [52]. Caffeine is a potent neurostimulant and also has neuroprotective effects. Its effects on the CNS arise through the competitive non-selective inhibition of adenosine A1 and A2A receptors. Epidemiological studies suggest that consumption of 3-4 cups of coffee daily (~200mg) can reduce the risk of PD [53, 54].
This is further supported by randomized trials and in-vitro studies that highlight caffeine’s ability to antagonise adenosine receptors, in particular A2A receptors found in striatopallidal neurons. Blockade of A2A receptors improves mobility in PD patients [55, 56].
Blockade of adenosine receptors improves motor functions of PD marginally in both animals and humans. Moreover, long-term exposure to caffeine in hemiparkinsonian mice may alter L-dopa responses in PD via unknown mechanisms [57]. The benefits of caffeine and its use in adjuvant therapy are outlined in the rest of this literature review.
3. DIFFERENT EFFECTS OF CAFFEINE IN PD
3.1. Adenosine A2A Receptors
Adenosine A2A receptors are distributed in the BG, which is heavily innervated by dopaminergic neurons. Adeno-sine is a ligand for all adenosine receptors and at low concentrations it shows a greater affinity for A1 receptors where it decreases glutamate release from striatal neurons. At high concentrations, adenosine has a higher affinity for A2A receptors and exerts excitatory effects through stimulation of glutamate and Ach release in the striatum. Activation of adenosine A2A receptors also enhances GABA-A mediated inhibitory effects on the GPe [58].
Adenosine A2A receptors are primarily expressed on striatopallidal neurons and form functional heteromeric compl-exes with D2 receptors and metabotropic glutamate receptors [mGLU5]. In so doing, A2A receptors can control functions of indirect efferent pathways of the BG. It is important to note that A2A receptors are functionally equivalent to D2 receptors since they form heteromers with D2 receptors by interacting with the arginine-rich domain on D2 receptors. A similar interaction is also seen between D1-NMDA receptors [N-methyl-D-aspartate receptors] heteromerisation [59, 60] .Upon forming a heteromeric complex with D2 receptors, A2A and D2 receptors become reciprocal antagonists and form the basis of motor control, learning, motivation, and reward systems. D2 receptors normally antagonise A2A receptors’ activity by inhibiting adenylyl cyclase activation. However, when D2 receptor interacts with A2A receptor in heteromeric configuration, A2A receptor can reduce the binding affinity of dopamine to D2 receptor, preventing inhi-bition of motor activity [60]. In addition, co-stimulation of A2A with mGLU5 enhances A2A receptors ability to inhibit D2 receptors as well as to override the inhibitory tone imposed by endogenous dopamine via D2 receptors [61]. The underlying antagonistic properties of A2A receptor on D2 receptors with deficits in dopamine levels translates to akinesia, which is commonly seen in PD [61]. Moreover, loss of striatal dopamine in PD results in unopposed adenosine stimulation resulting in hyperactivity of indirect pathway and motor dysfunction, which are also features observed in PD.
3.1.1. Caffeine as an Adenosine A2A Receptor Antagonist
Caffeine is a potent neurostimulant and might protect against a number of neurodegenerative disease and may affect cell cycle function in certain types of cancer [62, 63]. Nevertheless, the role of caffeine in cardiovascular diseases remains inconclusive according to a prospective cohort study [64]. Emerging evidence from in vivo and in vitro studies suggests that caffeine may protect against development of PD by keeping the BBB intact, which is essential for neuronal survival and CNS functioning [65, 66]. In addition, caffeine may improve motor functions in PD patients by modulating adenosine A2A receptors at different dosages [10]. Caffeine is structurally similar to adenosine and is a non-selective antagonist at A1, A2A, A2B and A3 adenosine receptor subtypes [67]. A2A receptors are predominately found on striatopallidal neurons and are antagonised by caffeine, which results in decreased intracellular cAMP levels and a diminished release of GABA in the GP [67]. A2A antagonists like caffeine thus may decrease the release of inhibitory neurotransmitters [GABA] and increases excitatory activity [serotonin, noradrenaline] in the striatopallidal neurons by stimulation of dopamine D1 and D2 receptors, promoting motor activities in rats [68]. Nevertheless, rapid tolerance to the effects of caffeine develops in long-term use [69]. Animal studies by Popoli et al. suggests that chronic caffeine administration combined with a selective A2A receptor antagonist such as SCH 58261 results in less tolerance towards adenosine A2A receptor antagonists compared to when adenosine A2A receptor antagonists are administered on their own [70].
Several studies have reported a reduction in MPTP-induced destruction of dopaminergic neurons in mice administered with oral A2A receptor antagonists. This protected against the loss of nigral dopaminergic neuronal cells induced by 6-hydroxydopamine in mice. A2A antagonists also prevented the functional loss of dopaminergic nerve terminals in the striatum and the ensuing gliosis caused by MPTP in mice [71]. The mechanism underlying the neuroprotective effects of A2A antagonists such as caffeine is still not fully understood.
On the contrary, some studies have reported that the activation of A2A receptors may protect tissues from reperfusion injury since A2A receptors can inhibit release of pro-inflammatory cytokines [TNF-α, IL-6, IL-8 and IL-12] and increase the release of anti-inflammatory cytokines such as IL-10 [72-74]. Moreover, A2A receptor activation can reduce the release of a number of ROS from neutrophils and also reduces neutrophilic infiltration into endothelial cells.
In light of the contradictory findings reported in the literature, further evaluation of the role that A2A activation and antagonism plays in neuroprotection is needed.
3.2. Astrocytes and Their Role in PD
Aside from being an A2A antagonist, caffeine may help stabilise the blood-brain barrier by regulating astrocytes and inflammatory processes in animal models [66]. Astrocytes are glial cells found in the mammalian brain that play a pivotal role in maintaining various neuronal functions by transporting nutrients and metabolites to neurons through a number of transporters such as the malate-aspartate shuttle. There are two types of astrocytes found in substantia nigra: [75].
- Protoplasmic astrocytes which encapsulate neuronal bodies and synapses
- Fibrous astrocytes which interact with oligodendroglia and node of Ranvier
Research on astrocytes in mice has suggested that α-synuclein affects only protoplasmic astrocytes but not fibrous astrocytes [76].
Astrocytes are significant players in the central nervous system as they develop and maintain the BBB and promote neurovascular coupling. One extreme of astrocytes faces endothelial cells to maintain BBB and control cerebral blood flow [CBF] while the other extreme is in communication with numerous neurons [77]. Moreover, astrocytes can release chemokines, gliotransmitters and help in the uptake of glutamate and GABA from the synaptic cleft. Other functions of astrocytes include pH regulation, K+ buffering, metabolising dopamine, and producing antioxidants such as gluthathione [GSH] and superoxide dismutases [SOD 1, and 2] [78].
In neurodegenerative diseases such as PD, astrocyte functions are compromised. Neurons are more susceptible to injury due to their limited capacity to counteract free radical-induced damage. Furthermore, neurons require a number of nerve growth factors [NGF] which are derived from astrocytes [79]. In the absence of astrocytes, neuronal functions are compromised and cell death slowly ensues during brain degeneration in PD [79]. In addition, during the process of reactive astrogliosis in the substantia nigra [an abnormal increase in astrocyte numbers secondary to neuronal death], astrocytes release a number of proinflammatory cytokines that act on neighbouring neurons. The increased production of ROS and activation of apoptotic mechanisms is often observed in PD, and may lead to death of dopaminergic neurons. Reactive astrogliosis is one of the main features seen in astrocytes following brain injuries, and in neurodeg-enerative diseases caused by trauma, α-synuclein aggregation, and ischemia [79]. A number of researchers have suggested that neurotoxins such as MPTP are capable of inducing astrogliosis and microgliosis which are often accompanied by mitochondrial dysfunction, nuclear karyolysis, and eventually neuronal death [80].
It is widely reported in a number of animal studies that astrocytes actively participate in the death of dopaminergic neurons by activating apoptosis through cytokines [TNFα, IL-1B and IL-6] and nitric oxide [NO] [28]. NO is believed to promote the release of cytochrome C from dysfunctional mitochondria while cytokines bind to specific receptors in dopaminergic neurons to activate the pro-apoptotic caspase pathway [81, 82]. Oxidative damage is a common feature observed in PD brain samples [83]. The increased uptake of α-synuclein by astrocytes leads to up-regulation of inflammatory molecules causing further production of ROS which may then lead to DNA damage [84]. Further depletion of GSH and SOD 1 and 2 may facilitate production of ROS and reactive nitrogen species [RNS] in astrocytes. This then leads to structural alteration and aggregation of normal α-synuclein proteins [85, 86]. Recent findings in mice models of PD have found that the nitration (nitrogen group is introduced to tyrosine residue on α-synuclein) of α-synuclein induced by oxidative injury (activation of induced nitric oxide synthase [iNOS] and focal adhesion kinase [FAK]) can enhance α-synuclein fibril formation causing cell death [87, 88]. Therefore one may conclude that inhibiting glial inflammatory processes could reduce neuronal damage during PD [89].
3.2.1. Caffeine Inhibits Astrocyte-induced Inflammation in PD
Even though caffeine can antagonise all forms of adenosine receptors it remains ineffective in antagonising purinergic-7 receptors [P2X7R] which are ligand-gated cation channels that open in response to ATP binding and lead to cell depolarization. P2X7 receptors are present on neurons and astrocytes throughout the CNS and PNS [90]. Recent studies suggest that P2X7R mediates platelet aggregation as well as microglial activation during inflammation. Moreover, P2X7R are also responsible for mediating apoptosis [91]. The use of a P2X7R antagonist, in combination with an adenosine receptor antagonist such as caffeine may reduce the inflammatory processes that are triggered by both receptors and their subtypes [92].
In addition, a number of studies in newborn rat brain cultures suggest that high doses of caffeine can reduce glial cell proliferation. This may be useful in reducing the inflammatory process in PD brains [93]. By actively preventing glial cell proliferation, specifically astroglyosis, the progression of neurodegeneration may be stalled due to mecha-nisms that prevent the breakdown of the BBB and the excessive release of gliotransmitters such as glutamate that lead to neurotoxicity.
Caffeine is also reported to suppress the production of TNF-α through protein kinase A dependent mechanisms in human blood according to Horrigan et al. [94]. Furthermore, caffeine is reported to mediate glutathione synthesis, reducing the ROS and RNS induced inflammatory response in the hippocampus of rodents leading to neuroprotection [95].
Astrocytes also express adenosine A2A receptors that may be antagonised by adenosine A2A antagonists, favouring the increased uptake of glutamate into astrocytes and thus reducing glutamate levels at the synaptic cleft. This can reduce the incidence of neurotoxicity induced by the excessive stimulatory effects of neurotransmitters [95, 96]. In addition to increased gliotransmitter uptake, chronic caffeine intake in non-habitual caffeine consumers may reduce CBF acutely through an unknown mechanism, which may involve vasoconstriction of cerebral vessels [97, 98].
3.3. Overexpression of Cav 1.3 Ca2+ Channels and Inhibition by Caffeine in PD
Voltage-gated calcium channels 1.3 [Cav 1.3 Ca2+] are L-type calcium channels that are selectively permeable to calcium ions. Cav 1.3 Ca2+ channels have an α1 subunit which is a prime target for calcium-channel blockers such as the dihydropyridines [99]. In addition to astrocyte dysregulation, the activity of Cav 1.3 Ca2+ channels has been implicated in the neurodegeneration of dopamine-producing neurons in PD. Dopaminergic neurons found in SNc continuously generate low frequency basal activity using Cav 1.3 Ca2+ channels. These L-type calcium channels have low expression levels [100]. Other L-type calcium channels found in SNc include Cav 1.2 Ca2+ channels. These are exclusively expressed in dopaminergic neurons located in the SNc and are only activated at higher membrane potentials. Conversely, Cav 1.3 Ca2+ channels are activated at sub-threshold membrane potentials, making them ideal pacemaker cells in neurons. However, they require significantly more ATP to sustain their transient activity. Moreover, Cav 1.3 Ca2+ channels generate oxidative stress mediated by mitochondria during periods of pacemaker activity via unknown mechanisms [101].
Recent studies suggest that there is an increased density of Cav 1.3 Ca2+ channels in dopaminergic neurons in mice models of PD [102]. The increased expression of dopaminergic neurons in Cav 1.3 Ca2+ channels generates more ROS that leads to neurodegeneration of dopaminergic neurons as seen in PD. These changes in the expression of voltage gated calcium channel subtypes in PD are of particular therapeutic interest [102]. A number of studies have reported the potential benefits of dihydropyridine [DHP] calcium channel blockers in reducing pathogenesis of PD as well as mortality in elderly PD patients [103]. Other studies have reported that caffeine can inhibit ryanodine receptors [RyRs] in certain parts of the body [104]. RyRs play a central role in microglial-mediated inflammation, and are believed to contribute to pathogenesis of neurodegenerative disorders such as PD. Inhibition of RyRs shows therapeutic promise in the management of PD [105]. Studies have also shown that protein kinase C [PKC] activation may lead to phospho-rylation of serine residue 81 on the amino-terminus of Cav 1.3 Ca2+ channels, leading to their inactivation [106]. However, no studies have reported possible effects of caffeine on protein kinase C pathways in the CNS or on inhibition of Cav1.3 Ca2+ channels.
3.4. Efficacy of Caffeine in Different People and Genders
Caffeine may show promise in the pharamcotherapy of PD since it antagonises adenosine receptors, inhibits the inflammatory response, and possibly inactivates calcium channels. The effect of caffeine was reported to be dose-dependent in a randomised controlled trial showing that a daily dose of 100-200mg of caffeine can reduce the risk of PD motor symptoms, while a daily dose of above 200mg caffeine was ineffective [107]. The reason behind this discrepancy includes genetic variation and gender differences. Genetic variation in cytochrome CYP1A2 gives rise to fast and slow metabolisers of caffeine. CYP1A2 is a primary enzyme responsible for metabolism of 95% of caffeine into three active metabolites: paraxanthine, theophylline, and theobromine. Each of these metabolites exert different effects. Certain variations in CYP1A2 gene result in a reduced ability to metabolise caffeine slow metabolisers [108]. These individuals are prone to hypertension as high levels of caffeine in serum can activate systemic epinephrine release. Moreover, sustained hypertension leads to vascular disease and possibly early myocardial infarction and cerebro-vascular accidents, that may then induce secondary neurodegenerative disorders such as vascular parkinsonism [109, 110].
Conversely, fast metabolisers overexpress CYP1A2. This results in rapid and efficient metabolism of caffeine [108]. These individuals may require higher doses of caffeine in order to achieve the same therapeutic effects of caffeine in reducing the prevalence of PD. However, conflicting studies have suggested that slow metabolisers are not necessarily at higher risk of developing PD when compared to fast metabolisers and this warrants further long-term clinical trials [108].
Apart from variation in caffeine metabolism, gender may also account for differences in the incidence of PD and response to caffeine. PD is more prevalent in males than in females due to differences in physiological adaptation as well as differences in toxicant exposure rate [111]. Nevertheless, in a number of epidemiological studies, it is reported that male caffeine users were more likely to have lower incidence of PD compared to female caffeine users. This lead to a number of large prospective studies which highlighted the possible link between exogenous oestrogen in Hormone Replacement Therapy [HRT] and reduced efficacy in postmenopausal women with higher PD incidence [112, 113]. It seems that exogenous oestrogen present in HRT reduces half-life of caffeine similar to those seen in fast metabolisers of caffeine, suggesting that exogenous oestrogen may put females on HRT at a greater risk of developing neurode-generative diseases such as PD. In vitro animal studies have compared the effects of a 20mg caffeine dose administration in normal vs ovariectomised mice exposed to MPTP. Ovariectomised mice had reduced parkisonian symptoms compared to normal mice. In addition, when male mice were injected with exogenous oestrogen and MPTP, they showed the same parkinsonian symptoms as female mice with ovaries. This suggests that administration of exogenous oestrogen may somehow inhibit the neuroprotective effects of caffeine [114, 115].
3.5. Caffeine as an Adjuvant Therapy in PD
The evidence outlined above highlights a number of beneficial effects of caffeine that could possibly justify its use as an adjuvant drug in the management of PD. There are reported experimental uses of caffeine in treating PD. For instance, trihexyphenidyl [THP] is an anti-parkinsonian agent that has been clinically used to treat PD [116]. It is a potent M1 receptor antagonist that inhibits M1 receptors in different parts of the cerebral cortex. Its use is accompanied by side effects that include memory impairment [116]. When THP is co-administered with low doses of caffeine in mice, there is increased THP potency without memory impairment. Observational studies on reserpine-induced hypokinesia in rats reported no reversal of symptoms when caffeine or THP were used on their own [117, 118]. Conversely, when caffeine and THP were co-administered, they restored locomotion in reserpine-treated rats.
The authors concluded that low doses of caffeine co-administered with THP might help reduce a number of motor deficiencies in animals, which are seen in PD. Nevertheless, long-term randomized clinical trials are needed to assess such claims and therapeutic benefits of such co-administration [116].
Other studies have investigated the effects of caffeine co-administration with bromocriptine. This improves a number of motor functions in rats under an observational study. Bromocriptine is a dopamine agonist commonly used to treat PD. When Bromocriptine is administered individually in denervated rats, subthreshold motor activities were recorded [119, 120]. When Bromocriptine was co-administered with caffeine, levels of motor activity were slightly higher than with the isolated use of Bromocriptine. Nonetheless, tolerance to caffeine was reported in rats administered only caffeine for a significant period of time but no tolerance to Bromocriptine was reported. In addition, no tolerance to chronic use of caffeine was reported when it was co-administered with Bromocriptine, and sustained levels of motor activity was observed in these rats [120]. Therefore, it is important to explore the relationship between caffeine and other parkinsonian drugs to determine the underlying mechanisms that allow caffeine to enhance efficacy and reduce drug tolerance [120].
A number of novel approaches have been adopted by the scientific community in an attempt to find better and more efficacious treatment for PD. Currently, Parkinsonian symptoms are treated through medications such as L-dopa and dopamine agonists like pramipexole and rotigotine, which themselves are accompanied by significant side effects that range from hypotension to gastrointestinal disturbances [121, 122] . Moreover, most of these drugs undergo first pass metabolism through the liver and are metabolised by circulating enzymes such as Monoamine oxidases [MAO] which reduces their bioavailability in the body. Hence they do not offer a long-term solution in treating PD.
In summary, caffeine is a cheap and widely-available neuromodulator which can be used in the treatment of PD in conjunction with current therapy. Nevertheless the precise dosage and safety of caffeine is of particular concern especially when it is administered at high doses for a significant period of time; a number of adverse effects such as resting tremors have been reported in chronic caffeine users. Therefore, more long-term double-blinded randomized controlled trials in humans are necessary in order to evaluate the efficacy and possible uses of caffeine in treating conditions like PD.
CONCLUSION
It was not until recently that the scientific community began to notice the neuroprotective effects of caffeine following the observation of lower incidences of PD reported in chronic caffeine users. Recent animal and human studies have suggested that caffeine may help to treat PD since it can antagonise the adenosine A2A receptors and inflammatory changes in the BG. Moreover, caffeine can prevent apoptotic cell death by lowering caspase-3 activity that is induced by neurotoxins such as MPP+, as well as decrease the number of apoptotic fragmented nuclei. In doing so it may halt dopaminergic neuronal destruction and act as cytoprotective agent [123].
As a result, caffeine might be able to prevent neurotoxicity induced by LBs and LNs. The beneficial functions of caffeine may not be limited solely to neuronal cells as it may be able to decrease the activity and proliferation of non-neuronal cells like astrocytes, which are primary regulators of the BBB and produce a number of inflammatory molecules in early and late stages of PD. Further randomised trials are needed to support this hypothesis.
The physiological role of caffeine may also help reduce the incidence of drug tolerance that often develops in advanced stages of PD where patients are given higher dosage to achieve the same therapeutic effects. Nevertheless, the beneficial effects of caffeine are not observed in all individuals due to genetic and gender differences. Individuals with variants in CYP1A2 are known to metabolise caffeine at a much slower rate than normal, this potentially predisposes them to cardiovascular diseases.
Aside for genetic variation, gender difference may also limit the neuroprotective effects of caffeine since females on HRT are not protected from neurodegenerative diseases even when caffeine is consumed chronically [124]. Physiological studies have suggested that exogenous oestrogen in HRT may inactivate neuroprotective effects of caffeine which may predispose females on HRT at higher risk of developing neurodegenerative disorders like PD [115]. In addition, most studies are currently conducted on animals, and may not have similar effects in humans.
Furthermore there is no single universally agreed dosage of caffeine which has been agreed by the scientific community. Doses used in most studies varied from 20mg to over 200mg [107, 125]. Moreover, long-term double-blinded randomised trials are needed to show the correlation between caffeine usage and low incidence of PD. Nonetheless, caffeine remains an interesting area of active research and may one day prove to be a cheap alternative to current PD treatment. In addition, a number of caffeine analogues are under investigation in clinical trials and have shown some promising results. Therefore, it is not long before caffeine will be used as an adjuvant therapy in treating neurodegenerative diseases like PD.
LIST OF ABBREVIATIONS
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

