All published articles of this journal are available on ScienceDirect.

Functional and Genomic Features of Human Genes Mutated in Neuropsychiatric Disorders
Abstract
Background:
In recent years, a large number of studies around the world have led to the identification of causal genes for hereditary types of common and rare neurological and psychiatric disorders.
Objective:
To explore the functional and genomic features of known human genes mutated in neuropsychiatric disorders.
Methods:
A systematic search was used to develop a comprehensive catalog of genes mutated in neuropsychiatric disorders (NPD). Functional enrichment and protein-protein interaction analyses were carried out. A false discovery rate approach was used for correction for multiple testing.
Results:
We found several functional categories that are enriched among NPD genes, such as gene ontologies, protein domains, tissue expression, signaling pathways and regulation by brain-expressed miRNAs and transcription factors. Sixty six of those NPD genes are known to be druggable. Several topographic parameters of protein-protein interaction networks and the degree of conservation between orthologous genes were identified as significant among NPD genes.
Conclusion:
These results represent one of the first analyses of enrichment of functional categories of genes known to harbor mutations for NPD. These findings could be useful for a future creation of computational tools for prioritization of novel candidate genes for NPD.
INTRODUCTION
Neuropsychiatric disorders (NPD) represent a large burden on global public health, in terms of the disability-adjusted life-years associated with them [1]. Taking into account the severity and chronicity of some of these disorders, global annual costs of NPD have been estimated at several trillion dollars [2].
For several NPD, particularly for neurological disorders, a large heritability for subtypes with Mendelian inheritance has been identified [3]. In the last years, several large efforts have been carried out to identify the causal genes for a large number of NPD [4]. Initially, classical genome-wide linkage studies, followed for fine-mapping and gene sequencing analyses, were used. Recently, genome-wide and exome sequencing studies [5] have generated a large number of causal genes for NPD [6]. Several available databases provide information for genes mutated in specific categories of NPD [7]. However, there is a lack of a global functional analysis of all genes that are known to harbor mutations for NPD. In the current work, we present a comprehensive catalog of genes mutated in neuropsychiatric disorders and we explore the genomic and functional features of those 300 genes.
METHODS
Identification of genes mutated in NPD was carried out by a combination of automatic and manual search strategies of the scientific literature and associated databases. Original articles were identified and data (such as first author, gene names, disorders and PubMed identifiers –PMIDs-] were extracted and stored. HUGO Gene Nomenclature Committee [HGNC] database [8] was used for identification of official gene symbols and names. DAVID server [9] was used for conversion of HGNC IDs to Ensembl Gene IDs. Ensembl BioMart [10] was used for retrieval of chromosome, band, gene start and end, gene size, transcript count and GC% data. The LiftOver tool of the University of California at Santa Cruz [UCSC] genome browser [11] was used to convert coordinates from hg38 to hg19 assemblies, hg19 was used because the latest available annotation for that genome version was more complete.
DAVID server (9) was used for functional clustering and enrichment analysis: Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways, Gene Expression, Chromosomal Location, Interpro domains, UCSC Transcription Factor Binding Sites [TFBS], and Gene Ontology [GO] terms. Babelomics [FatiGO] Server [12] was used for functional enrichment analysis: miRNA targets and KEGG pathways. For both programs, the option of comparing against the entire genome was chosen and a False Discovery Rate (FDR) approach was used for correction for multiple testing. A random sample of protein coding genes (from Ensembl database, N=300) was generated to analyze continuous variables (gene length, GC content and transcript counts), which were compared using a Mann-Whitney U test using the Stata 11 program (those variables presented a non-normal distribution).
Protein Protein Interaction (PPI) data were retrieved from the Human Interactome Project (Center for Cancer Systems Biology, Harvard University, USA). It consolidates different datasets: HI-II-14 and Lit-BM-13 [13], HI-I-05 [14]; Venkatesan-09 [15] and Yu-11 [16]. It led to 3482 interactions for 134 NPD proteins and 619 interactor proteins. VLOOKUP option in Excel 2013 was used for generation and integration of novel tables. Cytoscape 3.1 [17] was used for analysis and visualization of PPI networks. To facilitate PPI visualization, a subnetwork of highly connected proteins (>25 connections) was generated with the respective options in Cytoscape. A PPI network enrichment analysis was carried out with the SNOW tool [12], focusing on the following parameters: relative betweenness, connections and clustering coefficient. A list of druggable genes [18] was downloaded from the DGIdb database [19].
Sequences of the corresponding orthologous genes in Hominoids (chimpanzee, gorilla, orangutan and gibbon) were downloaded from the Ensembl database [20] and aligned using the MUSCLE alignment program [21]. Geneious software was used as a bioinformatics platform for all comparative analyses [22]. Two groups of genes were created: A group of proteins that are highly conserved between primates (>90% identity) and a second, less conserved group (<90% identity). Genes that have a unique gene structure in humans, compared with orthologues, were identified. Additionally, NPD genes that are located near or inside fragile regions of human X chromosome were recognized [23].
| Category | Feature | n (%) | p value | FDR |
|---|---|---|---|---|
| Chromosomal Location | Chromosome X | 45/294 (15.3) | 1,0E-11 a | 8,1E-9 |
| Gene Size | Gene Length | 0.0000 d | ||
| Transcriptional Complexity | Transcript count | 0.0000 d | ||
| Gene Expression (GNF_U133A) | Expression in Occipital Lobe | 88/294 (29.9) | 2,9E-11 a | 3,1E-8 |
| Gene Expression (GNF_U133A) | Expression in Prefrontal Cortex | 73/294 (24.8) | 4,6E-7 a | 4,9E-4 |
| Protein Domains (INTERPRO) | Ion Transport Domain | 14/294 (4.8) | 3,8E-8 a | 5,8E-5 |
| TF binding sites (UCSC) | SOX5 | 148/294 (60.5) | 1,2E-12 a | 1,4E-9 |
| TF binding sites (UCSC) | ZIC2 | 108/294 (36.7) | 1,7E-12 a | 2,1E-9 |
| TF binding sites (UCSC) | PAX6 | 191/294 (65.0) | 1,5E-11 a | 1,8E-8 |
| TF binding sites (UCSC) | NF1 | 141/294 (48.0) | 7,4E-10 a | 9,1E-7 |
| TF binding sites (UCSC) | POU3F2 | 189/294 (64.3) | 4,4E-7 a | 5,4E-4 |
| TF binding sites (UCSC) | EN1 | 174/294 (59.2) | 6,9E-7 a | 8,5E-4 |
| miRNA targets | hsa-let-7a | 21/300 (7.0) | 0.001 b | 0.03 |
| miRNA targets | hsa-mir-92b | 18/300 (6.0) | 0.001 b | 0.04 |
| miRNA targets | hsa-let-7g | 23/300 (7.7) | 0.0005 b | 0.02 |
| Category | Feature | n (%) | p value | FDR |
|---|---|---|---|---|
| Biological Process (GO) | Nervous system development | 76/294 (25.9) | 1,6E-23 a | 2,8E-20 |
| Biological Process (GO) | Transmission of nerve impulse | 39/294 (13.3) | 2,3E-18 a | 4,1E-15 |
| Cellular Component (GO) | Neuron projection | 43/294 (14.6) | 3,8E-24 a | 5,2E-21 |
| Molecular Function (GO) | Ion channel activity | 29/294 (9.9) | 2,1E-10 a | 3,1E-7 |
| Signaling Pathways (KEGG) | Wnt signaling pathway | 8/300 (2.7) | 0.0008 b | 0.03 |
| Signaling Pathways (KEGG) | Notch signaling pathway | 5/300 (1.7) | 0.0003 b | 0.01 |
| Signaling Pathways (KEGG) | Long-term potentiation | 5/300 (1.7) | 0.001 b | 0.04 |
| Signaling Pathways (KEGG) | MAPK signaling pathway | 11/300 (3.7) | 0.001 b | 0.03 |
| Protein-Protein Interaction Networks | Relative betweenness | 0.01 c | ||
| Protein-Protein Interaction Networks | Connections | 0.01 c | ||
| Protein-Protein Interaction Networks | Clustering Coefficient | 0.0007 c |
RESULTS
300 genes were identified as known to harbor mutations for NPD (Table S1). These genes belong to several functional categories, such as neurotransmitter receptors, ion channels, synaptic proteins, adhesion molecules, among other groups (Table S2). A functional enrichment analysis of these genes found several significant categories (Table 1). 15% of NPD genes are located on chromosome X and they have larger lengths and transcript counts.
In terms of functional pathways, genes related to Wnt, Notch, MAPK signaling and long-term potentiation mechanisms were overrepresented (Table 2). Among protein domains, only the ion transport domain from InterPro was significant. In terms of regulatory mechanisms, several transcription factors (TF) known to be involved in brain physiology and three miRNAs were identified (hsa-let-7a, hsa-mir-92b, hsa-let-7g) (Table 1), with an enrichment of genes expressed in prefrontal cortex and occipital lobe. A number of significant categories from the Gene Ontology were nervous system development, transmission of nerve impulse, neuron projection and ion channel activity (Table 2).
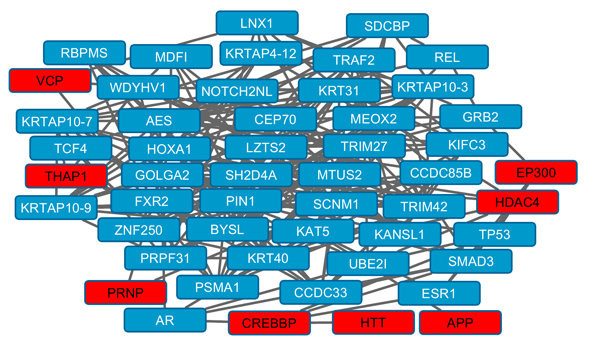
Several topographic parameters of protein-protein interaction networks were significant: Relative betweenness, connections and clustering coefficient (Table 2). Fig. (1) shows an overview of protein-protein interactions for a subnetwork of highly connected proteins. Sixty six NPD genes were identified as known as druggable (Table S3).
From the analysis of conservation among orthologues of NPD genes, two main groups were identified: A group of 272 genes that are highly conserved between primates (>90% identity) and a second, less conserved group (<90% identity) with 28 genes. A multiple alignment of the second group of orthologous genes showed that the encoded proteins had from 55.1 to 90.6% identity, with a percentage of identical sites between 13.5 to 79.2% (Table S4). As an example, Fig. ( S1) shows the alignment of the REEP1 orthologous genes, highlighting their low protein identity and Fig. (S2) shows the protein alignment of ARID1B, underscoring that the human protein has 429 additional amino acids at the N-terminal position (1 to 429 aminoacids) compared with orthologous genes found in Hominoids. Finally, nine NPD genes, highly conserved in primates, were found inside or adjacent to fragile regions previously reported in the human X chromosome (Table S5).
DISCUSSION
These results represent one of the first analyses of enrichment of functional categories of genes known to harbor mutations for NPD [4]. Previous studies that were focused on analyses of all genes for human diseases identified several genomic features [such as gene length] that were significant predictors [24].
In this study, we found several genomic features for NPD, such as larger gene lengths and transcript counts, location on chromosome X, presence of ion transport protein domains, expression in prefrontal cortex and regulation by several transcription factors that are known to be involved in brain function [4, 25]. As miRNAs are being identified as novel major regulators of brain function and NPD [26], it is interesting that in this study we found a possible common regulation by three miRNAs. Given the large number of features tested, a false discovery rate approach was used for correction for multiple testing.
In terms of functional analyses, we found an enrichment of categories such as gene ontologies related to neural transmission and plasticity and signaling networks linked to synaptic plasticity (such as Wnt and Notch), which have been previously postulated as underlying several NPD [27-29]. Of special interest, from a systems biology perspective, we found several topographic parameters of protein-protein interaction networks that were significant for NPD genes [30, 31]. We found that 66 NPD genes are known to be druggable, a finding of relevance for development of novel therapeutic interventions [19].
We found that nine NPD genes are located inside or adjacent to fragile regions previously reported in the human X chromosome [23], with 28 NPD genes found to be less conserved among primates (<90% identity) and with 5 NPD genes showing a unique gene structure in humans, compared with orthologues.
Of special relevance, from a global public health perspective, is the future identification of additional causal genes for NPD, particularly in developing countries [32-36]. These results could be useful for the future creation of computational tools [37] that allow prioritization of novel candidate genes (including ncRNAs [26, 38]) for NPD, incorporating several of the parameters that were found in this work as significant for NPD genes.
ETHICAL APPROVAL
This article does not contain any studies with human participants or animals performed by any of the authors.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by research grants from VCTI-UAN (grant # 2016220) and Colciencias (grant # 823-2015). We thank Professor Jason Moore for his important suggestions.

