All published articles of this journal are available on ScienceDirect.

Cannabidiol Successful Therapy for Developmental and Epileptic Encephalopathy Related to CYFIP2
Authors Info & Affiliations
Abstract
Background:
The knowledge about the molecular basis of epilepsies has increased enormously with the advent of next-generation sequencing (NGS) technology, and CYFIP2 is one of the many genes recently recognized and associated with epilepsy. Pathogenic variants in CYFIP2 cause Developmental and Epileptic Encephalopathy 65 (DEE65), which is characterized by hypotonia, profound developmental delay, and epilepsy.
Case Presentation:
Herein, we report a 3-year-old male with an early onset epileptic encephalopathy (Ohtahara syndrome) evolving to Lennox-Gastaut syndrome refractory to several antiseizure medications. Whole exome sequencing (WES) disclosed a heterozygous pathogenic variant p.(Arg87Cys) in CYFIP2, which occurred as a de novo event. After the introduction of cannabidiol, the patient remained seizure-free for 16 months and had a marked electroencephalographic improvement.
Conclusion:
Cannabidiol might be a therapeutic option for CYFIP2-related epilepsy
1. INTRODUCTION
For many decades, the etiology of infantile-onset severe epilepsies remained obscure, and most of them were attributed to acquired injuries [1]. In the last two decades, advancements in molecular genetics allowed the identification of close to a hundred genes associated with what is now collectively known as developmental and epileptic encephalopathies [2, 3].
CYFIP2 (Cytoplasmic FMRP-interacting protein 2, OMIM * 606323) is one of the many genes that were recognized to be associated with infantile-onset severe epilepsy, leading to a condition named developmental and epileptic encephalopathy 65 (DEE65, OMIM # 618008). CYFIP2 gene encodes a component of the WASP-family verprolin-homologous protein (WAVE) regulatory complex (WRC) that, in combination with other proteins, have a crucial role in important neurodevelopmental processes, such as axon guidance and regulation of synapse morphology involved in actin dynamics, axon elongation, dendritic spine morphogenesis, and synaptic plasticity [4, 5].
The first individuals who have been reported involving the CYFIP2 gene had a de novo 5q33.3 to q35.1 deletion, 16Mb-long, and showed developmental delay, mild intellectual disability, minor facial abnormalities, seizures, and behavior problems [6-8].
In 2018, Nakashima et al. discovered four patients with de novo missense pathogenic mutations in CYFIP2 and early-onset epileptic encephalopathy, significant developmental delay, acquired microcephaly, facial morphological abnormalities, and hypotonia using trio-based whole-exome sequencing (WES) [5]. Initial interictal EEGs showed a burst suppression pattern, characteristic of Ohtahara syndrome and/or hypsarrhythmia typical of West syndrome. Brain MRI disclosed diffuse cerebral atrophy, especially of the frontal lobes [5]. These four patients had a de novo substitution at codon 87, replacing arginine for cysteine, proline, or leucine. The authors suggested that variants in this position may disrupt hydrogen bonding, leading to structural instability of WAVE regulatory complex with a gain-of-function, modified signaling pathway, and correlated with severe neurological disorders [5].
Zweier et al. (2019) reported 12 patients carrying a total of eight distinct de novo variants in CYFIP2 with a shared phenotype of intellectual disability, seizures, and hypotonia [9]. Morphological features shared among patients included high forehead, narrow, mildly up-slanting eyelid fissures, apparent hypertelorism, bulbous nasal tip, full cheeks, everted lip vermillion, and retrognathia [9]. Most of the patients showed generalized or truncal hypotonia, limb spasticity, visual impairment and/or strabismus. Brain MRI showed unspecific structural anomalies ranging from atrophy and hypomyelination. Epilepsy was untreatable in 6 of 12 patients, and many types of seizures were reported, including absences, myoclonic, generalized tonic or tonic-clonic, and epileptic spasms. Electroencephalogram disclosed variable epileptiform discharges, including focal, multifocal, and generalized, as well as burst-suppression and hypsarrhythmia [9].
Begemann et al. (2021) added 19 more patients with de novo CYFIP2 variants, eight of whom had epilepsy, six were refractory, and two were seizure-free with antiseizure medications (ASM) [10]. The electroencephalogram findings were mixed, with four individuals experiencing hypsarrhythmia [10]. The 16 individuals identified with likely disease-associated missense variants harbored 11 novels and two recurrent substitutions in CYFIP2. Three individuals had putative loss-of-function (LoF) variants of unknown significance. Missense variants of CYFIP2 are spatially clustered in the tertiary structure and are predicted to weaken the interaction with WASF1 or NCKAP1, leading to increased WASF1 activation [9, 10].
Some developmental and epileptic encephalopathies could be associated with pattern electro-clinical characteristics syndromes, such as Ohtahara syndrome, West syndrome, or Lennox-Gastaut syndrome (LGS) [3]. LGS is an electroclinical syndrome that includes multiple seizure types, intellectual disability and/or behavior disorders, and electroencephalogram with interictal diffuse 1.5 to 2.5 Hz slow spike-and-wave discharges during the awake state and paroxysmal fast activity during sleep. Optimal treatment for LGS remains uncertain, and studies have shown no drug to be highly efficacious [11]. For patients with newly diagnosed LGS, sodium valproate is the first-line treatment, but if it is ineffective, lamotrigine, rufinamide, topiramate, cannabidiol, and felbamate may also be useful as adjunctive treatment; clobazam might be useful for drop attacks. Nonpharmacological therapies, including the ketogenic diet, vagus nerve stimulation, and callosotomy, should be considered in children and young adults with pharmaco-resistant epilepsies LGS syndrome [12, 13].
Cannabidiol (CBD) is a non-psychoactive compound derived from the marijuana plant, which has been approved for the treatment of Dravet syndrome and Lennox-Gastaut syndrome and is thought to have broad antiseizure properties that might be beneficial for other types of intractable epilepsy or developmental and epileptic encephalopathy [14].
We describe a patient with a de novo pathogenic variant of CYFIP2 who had an early-onset epileptic encephalopathy (Ohtahara syndrome) that progressed to Lennox-Gastaut syndrome and became seizures free after using cannabidiol.
2. CASE REPORT
This 3-year-old male was born after an uneventful pregnancy and at full term, and he was discharged from the maternity ward in good health three days later. Parents were first-cousins. Epileptic spasms with a generalized onset were found at 20 days of birth, and an electroencephalogram showed a burst suppression pattern with high amplitude epileptiform discharges followed by diffuse flattening of the recording, which is characteristic of Ohtahara's syndrome. Many anti-seizure medications, such as phenobarbital 5 mg/kg/day, clobazam 1.0 mg/kg/day, valproate 50 mg/kg/day, and levetiracetam 40 mg/kg/day, had no therapeutic response. Additionally, a trial with pyridoxine (100/day) and pyridoxal phosphate (40 mg/kg/day for one week) was not effective. After the introduction of vigabatrin (increased until 180 mg/kg/day), there was a clinically significant reduction of epileptic spasms that was not accompanied by development or EEG improvement.
He was referred to an epilepsy center for investigation and clinical management at 11 months of age. Physical examination revealed minor facial abnormalities (Fig. 1), normal head circumference (10th percentile), global hypotonia with spastic tetraparesis, hyperreflexia, and ankle clonus. He developed a sleep disturbance that was managed with melatonin.
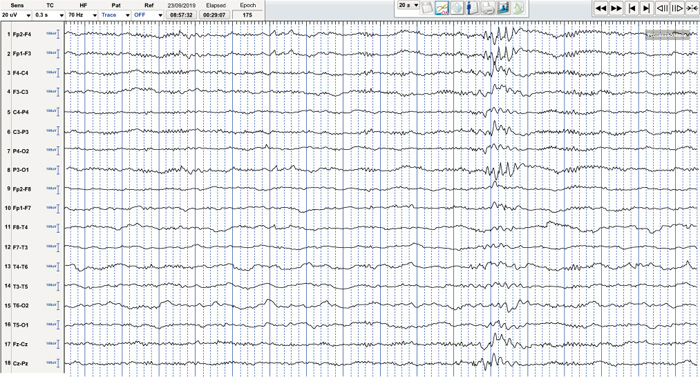
At the age of 14 months, his epileptic spasms stopped, and he began to experience several generalized tonic seizures, as well as some episodes of decreased awareness in both cephalic and ocular forms. A video-electroencephalogram revealed bitemporal slow spike waves (2-2,5 Hz), as well as electroclinical atypical absences and tonic seizures during sleep, all of which are compatible with LGS (Fig. 2). Topiramate (5 mg/kg/day) and then lamotrigine (6 mg/kg/day) were added to the treatment regimen, along with the gradual withdrawal of other ASM. There was a decrease in the number of atypical absences but no change in the number of tonic seizures.


Hundreds of tonic seizures without a trigger occurred daily, requiring re-hospitalization at 20 months of age, which was treated with intravenous phenytoin, phenobarbital, and midazolam, with partial seizure control. After three days, cannabidiol (CBD) (at a dose of 10 mg/kg/day) was related to progressive improvement and seizure-free status. When the EEG was performed three months after cannabidiol was introduced, it was normal when the subjects were awake, but it showed a bifrontal focal abnormality with generalization when they were sleeping. The metabolic workup was normal. Brain MRI at 4 months exhibited enlargement of the frontotemporal CSF space. An MRI a year later revealed diffuse cortical atrophy, especially in the frontotemporal region, as well as cerebellar white matter hyperintensity (Fig. 3) WES identified a de novo pathogenic variant in CYFIP2 [GRCh37Chr 5:15,6721,843C>T; c.259C>T ENST00000521420; p.(Arg87 Cys)]. Arginine at codon 87 is highly conserved among vertebrates, and its substitution for serine, proline, leucine, and cysteine is known to be pathogenic. Specifically, p.(Arg87Cys) have been reported and submitted to the variants repository many times, and it is a well-established pathogenic variant (https://www.ncbi.nlm.nih.gov/clinvar/variation/430807/).
He remained seizure-free with a severe developmental deficit after 16 months of cannabidiol usage. Currently, he is receiving vigabatrin (150 mg/kg/day), topiramate (9 mg/kg/day), and cannabidiol (10 mg/kg/day).
3. DISCUSSION
Different genes are associated with the etiology of developmental and epileptic encephalopathies, and some intellectual disability (ID) and epilepsy genes converge into common networks and play key roles in neurogenesis, neuronal migration, and synaptic functions [2, 3]. De novo variants in CYFIP2 are a cause of Developmental and Epileptic Encephalopathy 65 (DEE65), which is characterized by severe development delay; hypotonia; acquired microcephaly; facial morphological features; brain MRI abnormalities (cerebral and cerebellar atrophy, dysmorphic corpus callosum, hypomyelination or/and enlarged ventricles); epilepsy with many types of seizures and electroencephalogram discharges, including focal, multifocal, burst suppression pattern, and hypsarrhythmia [5, 9, 10]. Variants in CYFIP2 affecting the arginine at position 87 cause profound ID, early-onset refractory epilepsy, and hypotonia, leading to a severe phenotype [9]. Other CYFIP2 variants with suspected loss-of-function (LoF) variants show a milder phenotype, indicating that the complete loss of one allele product does not result in the severe neurodevelopmental phenotype seen with pathogenic de novo missense CYFIP2 variants [10].
In this patient, early-onset epilepsy with EEGs showed a burst suppression pattern, which was a characteristic of Ohtahara syndrome and pharmaco-resistant to therapy. Some ASM were used without measurable response, and others, such as vigabatrin and topiramate, were able to promote seizures partial control. However, the patient became seizure-free only after CBD was added-on. Zweier et al. described some seizure-free patients, but none met LGS criteria [9]. Zweier et al. reported two seizure-free patients, but none who met clinical-electroencephalographic criteria for LGS [9]. One stopped having seizures after taking levetiracetam and then switched to cannabidiol, while the other responded to lacosamide [5]. The majority of patients with a CYFIP2 variant required a combination of several ASM, including topiramate, levetiracetam, and valproate [5, 9, 10].
In this case, CBD was the most effective therapy for epilepsy. After its introduction, there was a significant clinical and electrographic improvement, which has been maintained for the last 16 months without side effects. Devinsky et al. (2018) published the first double-blind, placebo-controlled clinical trial using purified CBD for patients with LGS for drop seizures [15]. Adding 10 or 20 mg/kg/day of CBD to a conventional ASM led to greater reductions in the frequency of drop seizures than the lower doses used in this study. Nevertheless, no patient remained seizure-free throughout the whole study period (28 days) [15]. Cannabinoids' anticonvulsant properties, which act through various receptors and channels, can help control seizures and epilepsy. THC and CBD have been shown to diminish the duration, severity, and incidence of epileptic seizures and remote adverse effects [16]. This patient has a CYFIP2 gene mutation, which causes pharmacoresistant epilepsy. His clinics and EEG are consistent with LGS, a kind of epilepsy that has recently been shown to respond to cannabinol therapy.
CONCLUSION
Mutations in the CYFIP2 gene cause a phenotype of the development and epileptic encephalopathy spectrum, which includes intellectual disability, minor facial abnormalities, and epilepsy. We present a patient with a CYFIP2 p.(Arg87Cys) pathogenic variant who had epilepsy that was resistant to multiple medications and was eventually controlled with cannabidiol.
Our report suggests that cannabidiol is an option for epilepsy treatment in patients with developmental and epileptic encephalopathy 65 (DEE65, OMIM # 618008) and Lennox-Gastaut Syndrome.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This project was approved by the Institutional Ethical Committee (CAAE 09609319.1.0000.5269, Protocol number 4566159).
HUMAN AND ANIMAL RIGHTS
No animals were used in this research. All human research procedures were followed in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
Written informed consent was obtained from the parents of the patient for publication of this case report and any accompanying images.
STANDARDS OF REPORTING
The CARE guidelines and methodology have been followed.
FUNDING
None.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors are thankful to the team at the Fernandes Figueira Institute – Fiocruz in Rio de Janeiro, Brazil, for their assistance with patients and procedures.

